- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Identification of Insertion Site by RESDA-PCR in Chlamydomonas Mutants Generated by AphVIII Random Insertional Mutagenesis

Published: Vol 8, Iss 3, Feb 5, 2018 DOI: 10.21769/BioProtoc.2718 Views: 8819
Reviewed by: Trinadh Venkata Satish TammanaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Chlamydomonas reinhardtii is frequently used as a model organism to study fundamental processes in photosynthesis, metabolism, and flagellar biology. Versatile tool boxes have been developed for this alga (Fuhrmann et al., 1999; Schroda et al., 2000; Schroda, 2006). Among them, forward genetic approach has been intensively used, mostly because of the high efficiency in the generation of hundreds of thousands of mutants by random insertional mutagenesis and the haploid nature therefore phenotypic analysis can be done in the first generation (Cagnon et al., 2013; Tunçay et al., 2013). A major bottleneck in the application of high throughput methods in a forward genetic approach is the identification of the genetic lesion(s) responsible for the observed phenotype. In this protocol, we describe in detail an improved version of the restriction enzyme site-directed amplification PCR (RESDA-PCR) originally reported in (González-Ballester et al., 2005). The improvement includes optimization of primer combination, the choice of DNA polymerase, optimization of PCR cycle parameters, and application of direct sequencing of the PCR products. These modifications make it easier to get specific PCR products as well as speeding up subcloning steps to obtain sequencing data faster.
Keywords: Chlamydomonas reinhardtiiBackground
In addition to the restriction enzyme site-directed amplification PCR (RESDA-PCR) (González-Ballester et al., 2005), several other molecular techniques have been developed to identify insertion sites within the nuclear genome, including Genome Walker (Stirnberg and Happe, 2004), thermal asymmetric interlaced PCR (TAIL-PCR) (Dent et al., 2005), 3’-rapid amplification of cDNA ends (3’RACE) (Meslet-Cladiere and Vallon, 2012), Mme1-based insertion site sequencing strategy (ChlaMmeSeq) (Zhang et al., 2014), or whole-genome resequencing (Goold et al., 2016). RESDA-PCR is based on the use of specific primers of the marker gene combined with the use of degenerate primers that anneal with sequences of restriction sites highly and randomly distributed in the nuclear genome. RESDA-PCR is one of the most commonly used, is not too expensive and has been found to give the highest possibility in identifying the flanking sequence in our hands.
Materials and Reagents
- Falcon conical centrifuge tubes, 15 ml (Corning, catalog number: 430055 )
- Eppendorf tube, 1.5 ml and 2.0 ml, Eppendorf QualityTM (Eppendorf, catalog numbers: 0030120086 and 0030120094 )
- Petri dishes, 90 mm in diameter (Thermo Fisher Scientific, SterilinTM, catalog number: 101R20 )
- Sterilized toothpick (Fujian Fuhua, FUHUA FANGYUANTM, catalog number: 855 )
- Chlamydomonas reinhardtii mutants generated by random insertional mutagenesis (Cagnon et al., 2013)
- One ShotTM TOP10 Chemically Competent cell (Thermo Fisher Scientific, catalog number: C404010 )
- Zero BluntTM TOPOTM PCR Cloning Kit (Thermo Fisher Scientific, catalog number: 450245 )
- Taq DNA polymerase (5,000 U ml-1), 10x Standard Taq Reaction Buffer, dNTPs (New England Biolabs, catalog number: M0273S )
- Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: 472301 )
- PCR-grade H2O (Sigma-Aldrich, catalog number: W1754 )
- Ethanol (Sigma-Aldrich, catalog number: 34852 )
- KOD Xtreme Hot Start DNA polymerase, dNTPs (2 mM each), 2x Xtreme buffer (Merck, Novagen®, catalog number: 71842 )
- Agarose (Sigma-Aldrich, catalog number: A9539 )
- ExactLadder® DNA PreMix 2 log (OZYME, catalog number: OZYC002-500 )
- SYBRTM Safe DNA gel stain (Thermo Fisher Scientific, catalog number: S33102 )
- UltraPureTM 10x TAE buffer (Thermo Fisher Scientific, catalog number: 15558026 )
- Luria broth (Sigma-Aldrich, catalog number: L1900 )
- Kanamycin sulfate (Thermo Fisher Scientific, catalog number: 11815024 )
- NucleoSpin® Gel and PCR Clean-up Kit (MACHEREY-NAGEL, catalog number: 740609.10 )
- NucleoSpin® Plasmid Miniprep Kit (MACHEREY-NAGEL, catalog number: 740588.10 )
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E9884 )
- Tris base (Sigma-Aldrich, catalog number: T6066 )
- Hexadecyltrimethyl ammonium bromide (CTAB) (Sigma-Aldrich, catalog number: H9151 )
- Isopropanol (Sigma-Aldrich, catalog number: W292907 )
- Phenol:chloroform:isoamyl alcohol (25:24:1) (Sigma-Aldrich, catalog number: P3803 )
- Proteinase K (20.0 mg ml-1) (Sigma-Aldrich, catalog number: P6556 )
- Lysis buffer (see Recipes)
- 0.7 M NaCl
- 10% SDS
- 0.5 M Tris-HCl, pH 8.0
- 10 mM EDTA
- 0.7 M NaCl
- 5 M KCl (see Recipes)
- 10% CTAB (see Recipes)
- 10% DMSO (see Recipes)
- 1.0% agarose gel (see Recipes)
- 1x TAE (see Recipes)
- 70% ethanol (see Recipes)
Equipment
- Shaker (Eppendorf, New BrunswickTM, model: Innova® 44 , catalog number: M1282-0002)
- AccumetTM pH meter (Fisher Scientific, model: 3-in-1 Set, catalog number: 13-636-AE153 )
- PCR Thermal Cyclers (Thermo Fisher Scientific, Applied BiosystemsTM, model: 2720 , catalog number: ED000651)
- Agarose electrophoresis tank (Bio-Rad Laboratories, model: Mini-Sub® Cell GT, catalog number: 1704401 )
- Gel Doc XR System (Bio-Rad Laboratories, model: Gel DocTM XR+ , catalog number: 5838)
- FisherbrandTM Common bench-top vortexer (Fisher Scientific, catalog number: 02-216-125 )
- Bench-top centrifuge (Beckman Coulter, model: Allegra® 64R , catalog number: 367586)
- Bench-top incubator (Eppendorf, New BrunswickTM, model: S41i , catalog number: S41I230011)
- NanoDrop 2000 (Thermo Fisher Scientific, model: NanoDropTM 2000 , catalog number: ND-2000)
- Multisizer 3 Coulter counter (Beckman Coulter, model: MultisizerTM 3 , catalog number: 6605697)
Software
- SPSS program (version 19.0)
Procedure
- DNA extraction and quantification
- Cultivate cells of Chlamydomonas reinhardtii (around 0.5 x 106 cells ml-1) in shake flasks at 25 °C under constant white fluorescent light (100 µmol photons m-2 m-1) in TAP liquid media (Harris, 2001) with gentle shaking. Normally it will reach exponential phase (around 5 x 106 cells ml-1) after 24 h.
- Isolate and purify the genomic DNA of the mutant with the CTAB method (Schroda et al., 2001). Briefly, after cells grow to logarithmic phase (around 5 x 106 cells ml-1), harvest a total of 5 x 107 cells by centrifugation and suspend in 500 µl lysis buffer (100 mM Tris-HCl, pH 8.0, 1.75 mM EDTA, 150 mM NaCl, 2% [w/v] SDS, and 1.0 mg ml-1 Proteinase K).
- Incubate the lysates for 2 h at 55 °C, and then add 80 µl of 5 M KCl and 70 µl of preheated 10% CTAB and incubate for 10 min at 65 °C.
- Extract the lysates twice with (500 µl) phenol:chloroform:isoamyl alcohol (25:24:1, v/v/v), and then add (500 µl) chloroform to eliminate the remaining phenol.
- The nucleic acids are precipitated for 20 min on ice by addition of 500 µl of isopropanol, and washed with 70% cold ethanol.
- Resuspend the air-dried pellets in 50 µl sterile water.
- Quantify the purified DNA using NanoDrop 2000, and add water to adjust the final concentration to be of 50 ng µl-1.
Note: The concentration of the cells mentioned is measured with Multisizer 3 Coulter counter.
- Cultivate cells of Chlamydomonas reinhardtii (around 0.5 x 106 cells ml-1) in shake flasks at 25 °C under constant white fluorescent light (100 µmol photons m-2 m-1) in TAP liquid media (Harris, 2001) with gentle shaking. Normally it will reach exponential phase (around 5 x 106 cells ml-1) after 24 h.
- PCR amplification
This procedure largely follows the protocol first published by González-Ballester et al. (2005). To identify the DNA sequence around the site of insertion, two subsequent sets of PCR reactions are required, named here as the 1st and 2nd amplification. The 1st PCR amplification is to amplify a fragment from genomic DNA of the mutant using one AphVIII gene-specific primer with one of the degenerate primers (Figure 1). To increase the chances of amplification, 4 degenerate primers can be tested, and each of them contains a sequence-specific part (Q0 at the 5’ end) and a degenerate part (3’ end). The PCR product will be used as a template for a second round of PCR amplification (the 2nd PCR amplification) with the use of two sequence-specific primers. Degenerate primers (DegTaqI, DegPstI, DegAluI and DegSacII, including Q0) and the marker gene (AphVIII) specific primers (RB1 and RB4) are shown in Table 1.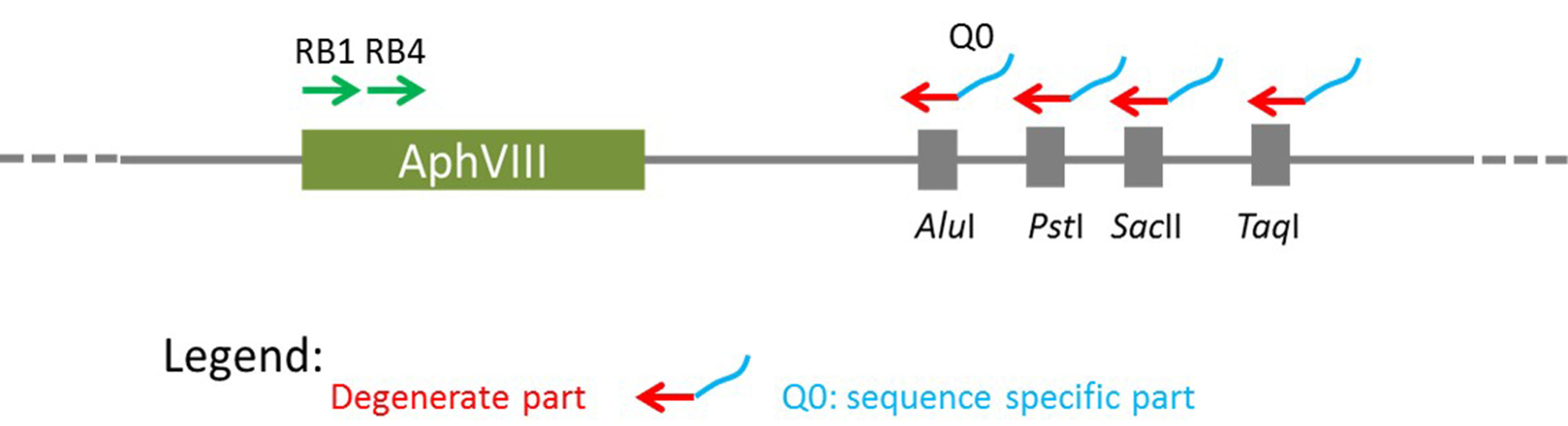
Figure 1. Outline of the principals for RESDA PCR. This schematic is adapted from González-Ballester et al., 2005.
Table 1. Primers and their sequences used in this protocol
★I, inosine, N, A + T + G + C; S, G + C; W, A + T.
*Gene-specific primers (RB1 and RB4) can be designed based on the type of marker gene (i.e., AphVII, Ble and aadA) used for mutagenesis.- The first amplification
In the 1st amplification, the AphVIII gene-specific primer (RB1) and one of the degenerate primers are used to amplify a fragment flanking the antibiotic marker gene AphVIII from the genomic DNA purified from the mutant. Four independent reactions can be set up by using four different degenerate primers and RB1, respectively. The composition of the PCR reaction mixture and cycling program are shown in Table 2 and Table 3, respectively.
Table 2. Composition of the PCR reaction mixture used for the 1st amplification
*Use one of the degenerate primers first.
Table 3. The PCR conditions used for the 1st amplification
*Each cycle contains thirteen steps sequentially; and totally twenty cycles were set up. - The second amplification
To get more specific PCR products, the maker gene-specific primer (RB4) and the specific primer (Q0) at the 5’ end of degenerate primers are used to amplify using the PCR from the 1st amplification as a template (One µl diluted PCR products (1/1,000) from 1st amplification). The composition of the PCR reaction mixture and cycling program are shown in Table 4 and Table 5, respectively.
Table 4. Composition of the PCR reaction mixture used for the 2nd amplification
Table 5. The conditions used for the 2nd PCR amplification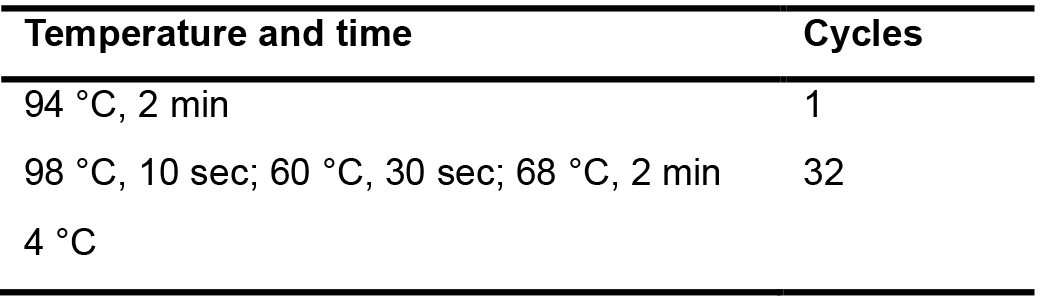
- The first amplification
- Agarose-gel electrophoresis and DNA fragments purification
- Separate the PCR products from the 2nd amplification on 1.0% agarose gel at 100 V for 30 min by electrophoresis, and then stain the gel with SYBR Safe (1/1,000) and take image with a GelDocXR System (Bio-Rad Laboratories). Some representative results are shown in Figure 2, and the examples of RESDA-PCR success rate after amplification by four independent degenerat primers and RB1, followed by further amplification with RB4 and Q0 primers are shown in Table 6.
- Cut–off the well-separated PCR products (marked with *) from the gel and purify using the NucleoSpin Gel and PCR Clean-up Kit (MACHEREY-NAGEL) according to manufacturer’s instructions.
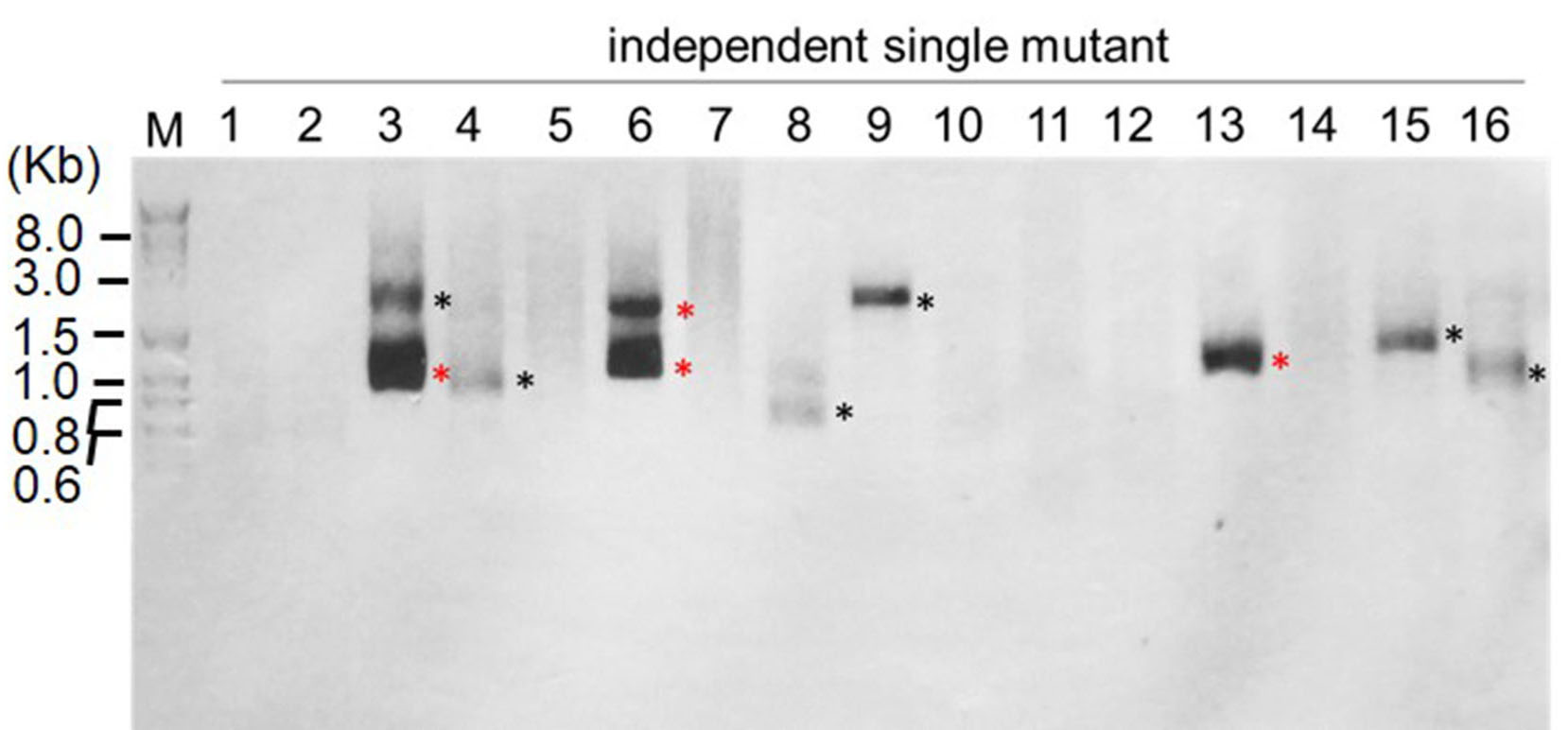
Figure 2. Examples of results of RESDA-PCR using the degenerate primer DegTaqI. The PCR products shown are the results of two rounds of PCR reactions: using DegTaqI and RB1, followed by further amplification with RB4 and Q0 primers.
Note: M refers to DNA marker (Exact Ladder DNA PreMix 2 log); and each lane (1, 2, 3…) refers to the amplification from independent single mutant. The black asterisk indicates the PCR fragment that will be cloned before sequencing, and the red asterisk indicates the PCR fragment that can be directly sequenced.
Table 6. Examples of RESDA-PCR success rate after amplification with four independent degenerat primers and RB1, followed by further amplification with RB4 and Q0 primers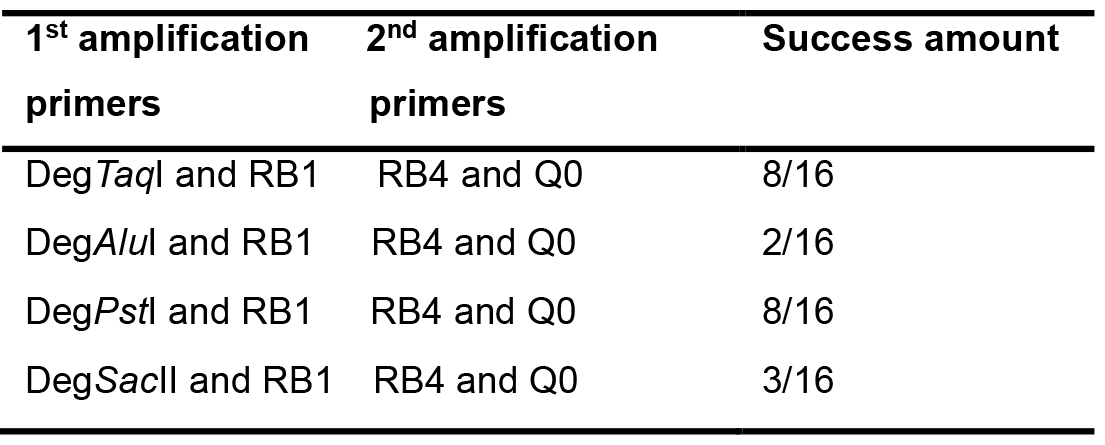
- Separate the PCR products from the 2nd amplification on 1.0% agarose gel at 100 V for 30 min by electrophoresis, and then stain the gel with SYBR Safe (1/1,000) and take image with a GelDocXR System (Bio-Rad Laboratories). Some representative results are shown in Figure 2, and the examples of RESDA-PCR success rate after amplification by four independent degenerat primers and RB1, followed by further amplification with RB4 and Q0 primers are shown in Table 6.
- Subsequent cloning of DNA fragment
Since the recovered PCR products generated by proofreading polymerases (KOD Xtreme Hot Start DNA polymerase) are blunt-end, in this protocol pCR II-Blunt-TOPO vector is used. Other Taq DNA polymerase can also be used in this step.- Directly ligate the recovered fragments to pCR II-Blunt-TOPO vector. This step can be completed within 5 min without adding a single deoxyadenosine (A) to the 3’ ends of PCR products. Add the ligation reaction components into a tube as described in Table 7, mix gently and incubate for 5 min at 22 °C.
Note: pCR-Blunt II-TOPO allows direct selection of recombinants via disruption of the lethal E. coli gene, ccdB (Bernard et al., 1994). Cells that contain a non-recombinant vector are killed upon plating. - The ligated products are transformed into One Shot competent cells (Thermo Fisher Scientific) according to manufacturers’ instructions. Transformants are selected on LB agar plates containing 50 μg ml-1 kanamycin (37 °C for overnight).
Table 7. Setup of the cloning reaction using the Zero BluntTM TOPOTM PCR cloning kit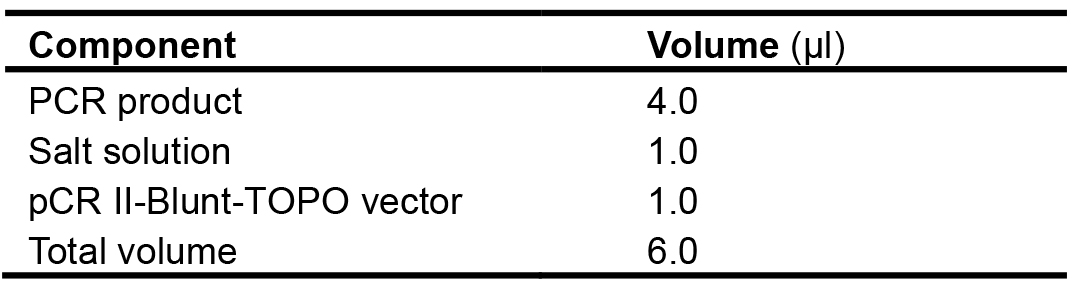
- Directly ligate the recovered fragments to pCR II-Blunt-TOPO vector. This step can be completed within 5 min without adding a single deoxyadenosine (A) to the 3’ ends of PCR products. Add the ligation reaction components into a tube as described in Table 7, mix gently and incubate for 5 min at 22 °C.
- Sequencing
- For each PCR fragment, culture three independent colonies separately in LB medium and extract plasmids using NucleoSpin Plasmid (MACHEREY-NAGEL) following manufacturer’s instructions.
- Sequence the purified plasmids using the T7 universal primer, and example of sequencing results for one representative mutant is shown in Figure 3.

Figure 3. Example of sequencing results for one representative mutant
Note: The partial sequences of AphVIII is underlined by red color and the sequences targeted to the genome of C. reinhardtii (v5.5) is underlined by black color, the sequences not underlined belong to the vector (pCR-Blunt II-TOPO) sequences.
- For each PCR fragment, culture three independent colonies separately in LB medium and extract plasmids using NucleoSpin Plasmid (MACHEREY-NAGEL) following manufacturer’s instructions.
Data analysis
The obtained sequences were firstly blasted against AphVIII expression cassette to confirm whether the RESDA-PRC fragment contains sequences of this cassette. If it is so, the obtained sequences were subsequently blasted against the genome of C. reinhardtii (v5.5 at Phytozome) (Merchant et al., 2007) to identify the flanking sequences around the cassette insertion site. The targeted genes of interest, i.e., maintenance-type DNA methyltransferase and acyl-CoA oxidase gene, were further studied by molecular genetics and biochemical analysis as we previously described (Kong et al., 2015 and 2017). All the experiments were performed in three biological replicates, and three plasmids of every single colony (containing PCR fragment) were sequenced, respectively. Statistical analysis was performed with SPSS program (version 19.0).
Notes
- This method is easily applicable to other insertional mutagenesis where marker genes other than AphVIII are used. Design of gene-specific primers is needed in this case.
- In 1st PCR amplification, it is essential to use Taq DNA polymerase (i.e., NEB) that has no proof-reading activity (lacking 3’ to 5’ DNase activity) to avoid the degradation of degenerate primers (containing inosine) hybridized to the genomic DNA. DMSO is used to reduce the hydrogen bonds in single-stranded DNA hairpin structures. We found out that the amplification with the DegPstI and DegTaqI degenerate primers had a higher probability of producing PCR products.
- In 2nd amplification, the diluted PCR product from the 1st PCR reaction at 1/1,000 was found to be the best situation to have higher chances of amplification. The PCR product often ranges from 0.8 kb to 1.5 kb. If the PCR product is specific with sufficient amount, it can be recovered and sequenced directly, i.e., without going through the step of cloning.
- pCR II-Blunt-TOPO vector is alternative for subsequent cloning of DNA fragment; it is also possible to use universal TA Cloning techniques for sub-cloning and DNA sequencing.
Recipes
- 0.7 M NaCl
Add 40.95 g NaCl crystalline (Sigma-Aldrich) to 800 ml sterilized MilliQ water and stir until NaCl is dissolved, and make the final volume to 1 L - 10% SDS
Dilute SDS powder (Sigma-Aldrich) with sterilized MilliQ water (1:10, w/v) to make the working solution - 0.5 M Tris-HCl, pH 8.0
Add 60.55 g Tris base power (Sigma-Aldrich) to 800 ml sterilized MilliQ water and stir until Tris base is dissolved, adjust the pH to 8.0 with HCl using pH meter to monitor the pH change and make the final volume to 1 L - 10 mM EDTA
Add 2.92 g Tris base power (Sigma-Aldrich) to 800 ml sterilized MilliQ water and stir until EDTA is dissolved and make the final volume to 1 L - Lysis buffer
Dilute 0.5 M Tris-HCl, pH 8.0, zss10 mM EDTA, 0.7 M NaCl, 10% SDS and Proteinase K (20.0 mg ml-1) to a mixture of the final concentration (100 mM Tris-HCl, pH 8.0, 1.75 mM EDTA, 150 mM NaCl, 2% SDS, and 1.0 mg ml-1 Proteinase K) - 5 M KCl
Add 372.76 g KCl crystalline (Sigma-Aldrich) to 800 ml sterilized MilliQ water and stir until KCl is dissolved, and make the final volume to 1 L - 10% CTAB
Dissolve CTAB powder (Sigma-Aldrich) with 0.7 M NaCl (10:100, w/v) - 10% DMSO
Dilute the DMSO (Sigma-Aldrich) with PCR-grade H2O (1:10, v/v) to the working solution - 1.0% agarose gel
Add agarose (Sigma-Aldrich) to 1 x TAE (1:100, w/v) - 1x TAE
Dilute UltraPure 10x TAE buffer (Thermo Fisher Scientific, UltraPureTM) with sterilized MilliQ water (1:10, v/v) to make the working solution - 70% ethanol
Dilute Ethanol (Sigma-Aldrich) with sterilized MilliQ water (7:3, v/v) to make the working solution
Acknowledgments
The work was supported by the French Agence Nationale pour la Recherche (ANR-Diesalg and ANR-MUsCA). This protocol is based on the protocol published by González-Ballester et al. (2005). There are no conflicts of interest.
References
- Bernard, P., Gabant, P., Bahassi, E. M. and Couturier, M. (1994). Positive-selection vectors using the F plasmid ccdB killer gene. Gene 148(1): 71-74.
- Cagnon, C., Mirabella, B., Nguyen, H. M., Beyly-Adriano, A., Bouvet, S., Cuine, S., Beisson, F., Peltier, G. and Li-Beisson, Y. (2013). Development of a forward genetic screen to isolate oil mutants in the green microalga Chlamydomonas reinhardtii. Biotechnol Biofuels 6(1): 178.
- Dent, R. M., Haglund, C. M., Chin, B. L., Kobayashi, M. C. and Niyogi, K. K. (2005). Functional genomics of eukaryotic photosynthesis using insertional mutagenesis of Chlamydomonas reinhardtii. Plant Physiol 137(2): 545-556.
- Fuhrmann, M., Oertel, W. and Hegemann, P. (1999). A synthetic gene coding for the green fluorescent protein (GFP) is a versatile reporter in Chlamydomonas reinhardtii. Plant J 19(3): 353-361.
- González-Ballester, D., de Montaigu, A., Higuera, J. J., Galvan, A. and Fernandez, E. (2005). Functional genomics of the regulation of the nitrate assimilation pathway in Chlamydomonas. Plant Physiol 137(2): 522-533.
- Goold, H. D., Nguyen, H. M., Kong, F., Beyly-Adriano, A., Legeret, B., Billon, E., Cuine, S., Beisson, F., Peltier, G. and Li-Beisson, Y. (2016). Whole genome re-sequencing identifies a quantitative trait locus repressing carbon reserve accumulation during optimal growth in Chlamydomonas reinhardtii. Sci Rep 6: 25209.
- Harris, E. (2001). Chlamydomonas as a model organism. Annu Rev Plan Physiol Plant Mol Biol 52: 363-406.
- Kong F., Liang Y., Légeret B., Beyly-Adriano A., Blangy S., Haslam R. P., Napier J. A., Beisson F., Peltier G. and Li-Beisson Y. (2017). Chlamydomonas carries out fatty acid β-oxidation in ancestral peroxisomes using a bona fide acyl-CoA oxidase. Plant J 90(2): 358-371.
- Kong, F., Yamasaki, T., Kurniasih, S. D., Hou, L., Li, X., Ivanova, N., Okada, S. and Ohama, T. (2015). Robust expression of heterologous genes by selection marker fusion system in improved Chlamydomonas strains. J Biosci Bioeng 120: 239-245.
- Merchant, S. S., Prochnik, S. E., Vallon, O., Harris, E. H., Karpowicz, S. J., Witman, G. B., Terry, A., Salamov, A., Fritz-Laylin, L. K., Marechal-Drouard, L., Marshall, W. F., Qu, L. H., Nelson, D. R., Sanderfoot, A. A., Spalding, M. H., Kapitonov, V. V., Ren, Q., Ferris, P., Lindquist, E., Shapiro, H., Lucas, S. M., Grimwood, J., Schmutz, J., Cardol, P., Cerutti, H., Chanfreau, G., Chen, C. L., Cognat, V., Croft, M. T., Dent, R., Dutcher, S., Fernandez, E., Fukuzawa, H., Gonzalez-Ballester, D., Gonzalez-Halphen, D., Hallmann, A., Hanikenne, M., Hippler, M., Inwood, W., Jabbari, K., Kalanon, M., Kuras, R., Lefebvre, P. A., Lemaire, S. D., Lobanov, A. V., Lohr, M., Manuell, A., Meier, I., Mets, L., Mittag, M., Mittelmeier, T., Moroney, J. V., Moseley, J., Napoli, C., Nedelcu, A. M., Niyogi, K., Novoselov, S. V., Paulsen, I. T., Pazour, G., Purton, S., Ral, J. P., Riano-Pachon, D. M., Riekhof, W., Rymarquis, L., Schroda, M., Stern, D., Umen, J., Willows, R., Wilson, N., Zimmer, S. L., Allmer, J., Balk, J., Bisova, K., Chen, C. J., Elias, M., Gendler, K., Hauser, C., Lamb, M. R., Ledford, H., Long, J. C., Minagawa, J., Page, M. D., Pan, J., Pootakham, W., Roje, S., Rose, A., Stahlberg, E., Terauchi, A. M., Yang, P., Ball, S., Bowler, C., Dieckmann, C. L., Gladyshev, V. N., Green, P., Jorgensen, R., Mayfield, S., Mueller-Roeber, B., Rajamani, S., Sayre, R. T., Brokstein, P., Dubchak, I., Goodstein, D., Hornick, L., Huang, Y. W., Jhaveri, J., Luo, Y., Martinez, D., Ngau, W. C., Otillar, B., Poliakov, A., Porter, A., Szajkowski, L., Werner, G., Zhou, K., Grigoriev, I. V., Rokhsar, D. S. and Grossman, A. R. (2007). The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318(5848): 245-250.
- Meslet-Cladiere, L. and Vallon, O. (2012). A new method to identify flanking sequence tags in Chlamydomonas using 3’-RACE. Plant Methods 8(1): 21.
- Schroda, M. (2006). RNA silencing in Chlamydomonas: mechanisms and tools. Curr Genet 49(2): 69-84.
- Schroda, M., Blocker, D. and Beck, C. F. (2000). The HSP70A promoter as a tool for the improved expression of transgenes in Chlamydomonas. Plant J 21(2): 121-131.
- Schroda, M., Vallon, O., Whitelegge, J. P., Beck, C. F. and Wollman, F. A. (2001). The chloroplastic GrpE homolog of Chlamydomonas: two isoforms generated by differential splicing. Plant Cell 13(12): 2823-2839.
- Stirnberg, M. and Happe, T. (2004). Identification of a Cis-Acting element controlling anaerobic expression of the Hyda gene from Chlamydomonas reinhardtii. Biohydrogen III: 117-127.
- Tunçay, H., Findinier, J., Duchene, T., Cogez, V., Cousin, C., Peltier, G., Ball, S. G. and Dauvillee, D. (2013). A forward genetic approach in Chlamydomonas reinhardtii as a strategy for exploring starch catabolism. PLoS One 8(9): e74763.
- Zhang, R., Patena, W., Armbruster, U., Gang, S. S., Blum, S. R. and Jonikas, M. C. (2014). High-throughput genotyping of green algal mutants reveals random distribution of mutagenic insertion sites and endonucleolytic cleavage of transforming DNA. Plant Cell 26(4): 1398-1409.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kong, F. and Li-Beisson, Y. (2018). Identification of Insertion Site by RESDA-PCR in Chlamydomonas Mutants Generated by AphVIII Random Insertional Mutagenesis. Bio-protocol 8(3): e2718. DOI: 10.21769/BioProtoc.2718.
Category
Plant Science > Plant molecular biology > DNA > Mutagenesis
Microbiology > Microbial genetics > Gene mapping and cloning
Molecular Biology > DNA > Genotyping
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.