- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Multiple Stepwise Gene Knockout Using CRISPR/Cas9 in Escherichia coli
(*contributed equally to this work) Published: Vol 8, Iss 2, Jan 20, 2018 DOI: 10.21769/BioProtoc.2688 Views: 28766
Reviewed by: David CisnerosAlba BlesaTomas Aparicio
Original research article
The authors used this protocol in:

Apr 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
With the recent implementation of the CRISPR/Cas9 technology as a standard tool for genome editing, laboratories all over the world are undergoing one of the biggest advancements in molecular biology since PCR. The key advantage of this method is its simplicity and universal applicability for species of any phylum. Of particular interest is the extensively studied Gram-negative bacterium Escherichia coli, as it is considered as the workhorse for both research and industrial purposes. Here, we present a simple, robust and effective protocol using the CRISPR/Cas9 system in combination with the λ Red machinery for gene knockout in E. coli. Crucial in our procedure is the use of a double-stranded donor DNA and a curing strategy for removal of the guide RNA encoding plasmid that allows starting a new mutation after only two working days. Our protocol allows multiple, stepwise gene knockout strains with high mutagenesis efficiencies applicable for high-throughput approaches.
Keywords: BiotechnologyBackground
The Gram-negative bacterium Escherichia coli is one of the most important organisms for biotechnological engineering. It has been successfully implemented in a wide range of processes in different sectors of industry, such as energy, agriculture, food production, biotechnology, and medicine. The biotechnology sector is improving fast due to a constant technological progress. In particular, the CRISPR/Cas9 technology is probably the biggest revolution in (molecular) biology since PCR (Ledford, 2015). Briefly, CRISPR/Cas9 protects bacteria from invasive genetic elements such as plasmids and viruses (Marraffini, 2015). Taking advantage of this kind of acquired immune system from prokaryotes, a remarkably powerful tool for genome editing based on the CRISPR/Cas9 systems has been developed (Jinek et al., 2012).
The CRISPR/Cas9 system consists of a guide RNA (gRNA) and the endonuclease Cas9 both forming a complex in a way that directs the enzyme to the target site that is complementary to the gRNA promoting site-specific cleavage (Sander and Joung, 2014). The enzymatically generated double strand break (DSB) is being subsequently repaired by the cell through either homologous recombination (HR) or non-homologous end joining (NHEJ) (Iyama and Wilson III, 2012). However, NHEJ works poorly in E. coli (Shuman and Glickman, 2007; Wright et al., 2016) and thus genome editing protocols cannot rely on such a mechanism for this Gram-negative bacterium. Therefore, most approaches aiming to modify chromosomal DNA in E. coli exploit the phage recombinase-mediated homologous recombination (recombineering) using either the Rac prophage system (Zhang et al., 1998; Datta et al., 2006) or the three bacteriophage λ Red proteins Exo, Beta, and Gam (Murphy, 1998; Muyrers et al., 1999; Ellis et al., 2001). In fact, the presence of the λ Red machinery considerably increases mutagenesis efficiency as demonstrated in a recent study (Pyne et al., 2015), especially when combined with the CRISPR/Cas9 technology (Jiang et al., 2013).
Our laboratory established a robust, simple and rapid protocol for multiple, stepwise gene knockout in E. coli using the CRISPR/Cas9 technology in combination with the λ Red machinery (Zerbini et al., 2017). Here, we want to provide our genome editing protocol in detail, which results in high mutagenesis efficiencies and has the potential to be applied in high-throughput approaches.
Materials and Reagents
- Pipette tips: GPR-10G (Mettler-Toledo, Rainin, catalog number: 17001862 ), GPR-250 (Mettler-Toledo, Rainin, catalog number: 17001861 ), GPR-1000 (Mettler-Toledo, Rainin, catalog number: 17001859 )
- 50 ml tube (Corning, Falcon®, catalog number: 352070 )
- Primo® Vacuum Filter Systems, 250 ml, 0.22 μm, PES (Euroclone, catalog number: EPVPE22250 )
- Syringe filters PES 0.22 µm filters (Euroclone, catalog number: EPSPE2230 )
- Petri dishes (Corning, Falcon®, catalog number: 351007 )
- MAX efficiencyTM DH5αTM competent cells (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18258012 )
Note: You can also use homemade competent cells of E. coli DH5α (see Step E1). - pCasRed plasmid (Zerbini et al., 2017), modified pCas9 plasmid from Addgene (Addgene, catalog number: 42876 ) available upon request
- pCRISPR-SacB plasmid (Zerbini et al., 2017), modified pCRISPR plasmid from Addgene (Addgene, catalog number: 42875 ), available upon request
- T4 Polynucleotide Kinase (PNK) (New England Biolabs, catalog number: M0201S )
- T4 Polynucleotide Kinase (PNK) Reaction Buffer (New England Biolabs, catalog number: B0201S )
- Oligonucleotide 1 or gDNA forward (Sigma-Aldrich; desalted, synthesis scale 0.025 µM) for cloning of the mutagenic pCRISPR-SacB-gDNA (see Procedure B)
- Oligonucleotide 2 or gDNA reverse (Sigma-Aldrich; desalted, synthesis scale 0.025 µM) for cloning of the mutagenic pCRISPR-SacB-gDNA (see Procedure B)
- T4 ligase (New England Biolabs, catalog number: M0202S )
- 10x T4 ligase buffer (New England Biolabs, catalog number: B0202S )
- 1 M NaCl (Sigma-Aldrich, catalog number: S7653-1KG )
- BsaI enzyme (New England Biolabs, catalog number: R0535L )
- DpnI enzyme (New England Biolabs, catalog number: R0176S )
- Alkaline phosphatase, Calf Intestinal (CIP, New England Biolabs, catalog number: M0290S )
- Wizard® SV Gel and PCR Clean-Up System (Promega, catalog number: A9282 )
- QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27106 )
- ATLAS ClearSight DNA Stain (BioAtlas, catalog number: BH40501 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266-1KG )
Note: See Recipes for preparation of the 100 mM working solution. - Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C3306-250G )
Note: See Recipes for preparation of the 100 mM working solution in 15% glycerol. - GoTaq Green Master Mix, 2x (Promega, catalog number: M7123 )
- LB broth Miller (Sigma-Aldrich, catalog number: L3522-1KG )
Note: See Recipes for preparation of LB medium. - LB agar (Sigma-Aldrich, catalog number: L2897-1KG )
Note: See Recipes for preparation of Petri dishes with LB-agar. - Kanamycin (Thermo Fisher Scientific, catalog number: 11815024 )
Note: Prepare a stock in a concentration of 50 mg/ml (see Recipes). - Chloramphenicol (Sigma-Aldrich, catalog number: C0378-25G )
Note: Prepare a stock in a concentration of 25 mg/ml (see Recipes). - L-(+)-Arabinose (Sigma-Aldrich, catalog number: A3256-100G )
Note: Prepare a stock in a concentration of 20% L-(+)-arabinose (see Recipes). - Sucrose (Sigma-Aldrich, catalog number: S5016-2.5KG )
Note: Prepare a stock in a concentration of 50% sucrose (see Recipes). - > 99% Glycerol (Acros Organics, catalog number: 158920010 )
Note: Prepare a 75% stock (see Recipes). - LB medium (liquid) (see Recipes)
- LB-agar (for Petri dishes) (see Recipes)
- Antibiotics (see Recipes)
- 20% L-(+)-arabinose (see Recipes)
- 50% sucrose (see Recipes)
- 75% glycerol solutions (see Recipes)
- Sterile 100 mM MgCl2 (see Recipes)
- Sterile 100 mM CaCl2 in 15% glycerol (see Recipes)
Equipment
- Pipettes: Pipetman P2L metal ejector (Gilson, catalog number: FA10001M ), Pipetman P10L metal ejector (Gilson, catalog number: FA10002M ), Pipetman P20L metal ejector (Gilson, catalog number: FA10003M ), Pipetman P200L metal ejector (Gilson, catalog number: FA10005M ), Pipetman P1000L metal ejector (Gilson, catalog number: FA10006M )
- pH meter (e.g., Fisher Scientific, model: Fisher ScientificTM accumetTM AB150, catalog number: 13-636-AB150 )
- -80 °C freezer for storage of strains (any commercially available freezer is suitable)
- -20 °C freezer (any commercially available freezer is suitable)
- Autoclave (e.g., Tuttnauer, model: 5075 EL , 160 L)
- Centrifuge, low temperature, up to 6,000 rpm (e.g., Thermo Fisher Scientific, Thermo ScientificTM, model: SorvallTM ST16 )
- DNA sequencer (or DNA sequencing service EUROFINS)
- Gene Pulser®/MicroPulserTM Electroporation Cuvettes, 0.1 cm gap (Bio-Rad Laboratories, catalog number: 1652089 )
- MicroPulserTM Electroporator (Bio-Rad Laboratories, catalog number: 1652100 )
- Incubator, temperature scale at least 30-37 °C, shaking range 0-300 r.p.m. (e.g., Eppendorf, New BrunswickTM, model: Innova® 44 )
- MilliQ water system (e.g., Elga Veolia, model: PURELAB® flex 3 )
- Spectrophotometer (e.g., Biochrom, model: WPA CO7500 Colorimeter )
- Thermocell mixing block (e.g., BionovaTec, model: MB 102 )
- Thermocycler for PCR (e.g., Eppendorf, model: Mastercycler® nexus )
- Transilluminator documentation system (e.g., UviTEC Cambridge, Laboratorium Life Science, model: UVIdoc HD5 )
Procedure
Generally, our CRISPR/Cas9 knockout protocol for Escherichia coli employs three main components:
1) pCasRed plasmid (Procedure A)
2) pCRISPR-SacB-gDNA plasmid (Procedure B)
3) The mutagenic double-stranded donor DNA oligonucleotide (Procedure C).
First, we transform with the pCasRed plasmid (1), which encodes not only the Cas9 endonuclease but also the λ Red machinery (see Procedure A for details). In a subsequent step, we then co-transform with the pCRISPR-SacB-gDNA plasmid (2) and the mutagenic double-stranded donor DNA (3).
Our protocol results in the inactivation of a target gene by directing the Cas9 endonuclease to the 5’-end of the target locus through a specifically designed guide RNA, encoded and previously cloned into the pCRSIPR-SacB-gDNA plasmid (see Procedure B for details). Upon DNA cleavage at the target locus, the mutagenic double-stranded donor DNA (ds-dDNA) allows for λ Red-assisted repair of the double strand break, thereby introducing an in-frame stop codon and removing the original protospacer adjacent motif (PAM) region (Figure 1). A workflow is shown in Figure 2.
When required, plasmid curing can be executed through sucrose selection, following Hale et al. (2010) (for other references see Gay et al., 1983; Steinmetz et al., 1983; Gay et al., 1985; Murphy et al., 2000). Briefly, the SacB gene encodes for levansucrase from Bacillus subtilis, which metabolizes sucrose into a toxic product if E.coli grows in the presence of this disaccharide. Therefore, cells will only grow if they have lost the pCRISPR-SacB-gDNA plasmid.
Concerning the length of deletion and the length of the ds-dDNA you may adapt this protocol for your own needs. Here, we generally describe 30 nucleotide deletions, corresponding to the target site using ds-dDNA of 70 base pairs. However, in our laboratory, we observed that longer deletions of up to 2,200 nucleotides work better with ds-dDNA of 120 base pairs (Zerbini et al., 2017).
Before you get started, you should start with the design of guides (Procedure B, Figure 3), donors and primers (Procedure C, Figure 4 and Figure 5, respectively). We recommend ordering them before moving to the wet laboratory procedures. We also recommend referring to the oligonucleotide sequences (target sites, donor DNAs, primers for colony PCR) published in our original research paper (Zerbini et al., 2017) if you want to include any positive control in your own experiments.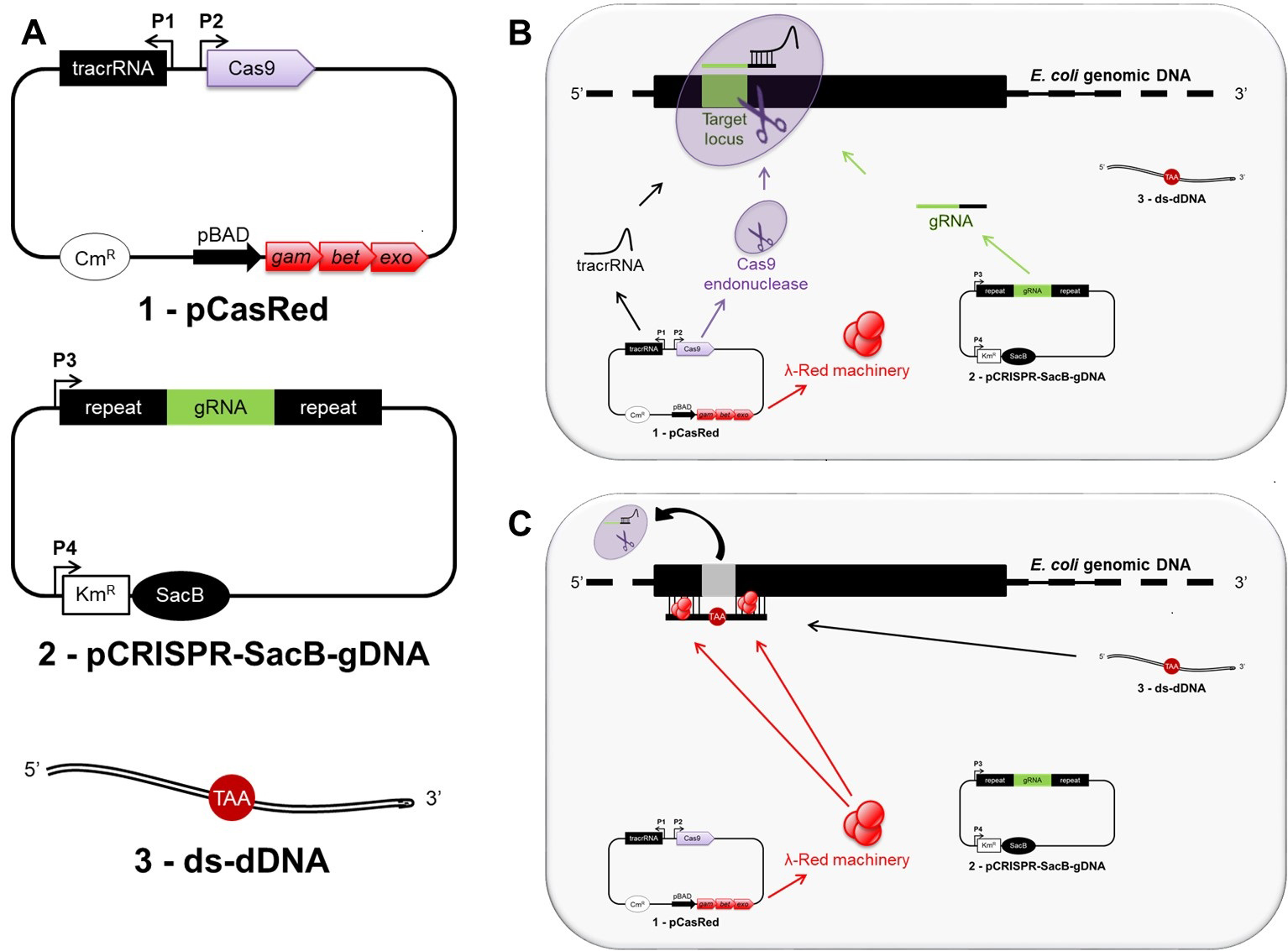
Figure 1. Overview of CRISPR/Cas9 genome editing strategy in Escherichia coli. A shows all three main components required for genome editing in E. coli: 1) the pCasRed plasmid expressing the λ Red (Exo, Beta, Gam) machinery, the Cas9 endonuclease, and the tracrRNA; 2) the pCRISPR-SacB-gDNA plasmid encoding the gRNA and the SacB gene; 3) a synthetic, double-stranded mutagenic oligonucleotide (ds-dDNA). After transformation, the tracrRNA anneals to the gRNA, which specifies the site of cleavage for Cas9 resulting in a three-component complex at the target locus, where the endonuclease activity mediates a chromosomal DNA double strand break (B). The double strand break is subsequently repaired by λ Red-mediated homologous recombination taking place between the extremities of the cleaved chromosomal DNA and the ds-dDNA (C).
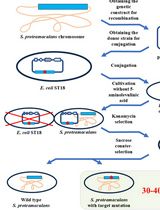
Figure 2. Schematic workflow for gene knockout in E. coli using the CRISPR/Cas9 technology. Step 1 includes the construction of the three main components needed for this protocol (see Figure 1). The E. coli strain you want to gene-edit (e.g., E. coli BL21(DE3)) has to be transformed with the pCasRed plasmid first (note asterisk, see Step E4a.i). The E. coli + pCasRed strain is the starting point for mutagenesis (Step 2), in which fresh electrocompetent cells are prepared (Step E3) and directly used for transformation (Step E4). The next day, a colony PCR is performed to identify positive knockout mutants (Steps E5 and E6), which are subsequently inoculated in liquid LB medium containing 5% sucrose for curing of the pCRISPR-SacB-gDNA used before (Step 3). Here, the protocol can be stopped or you continue with another round of mutagenesis following only the procedures from Step 2 highlighted in the dotted box. Legend: chloramphenicol (Cm), wild type (WT), positive mutant (+), escaper (-).
- Required plasmids
- The pCasRed plasmid (Zerbini et al., 2017) derives from the pCas9 plasmid from Addgene (#42876) and is available from our laboratory upon request.
Briefly, pCasRed encodes the Cas9 nuclease, the λ Red (Exo, Beta, Gam) cassette, the trans-activating CRISPR RNA (tracrRNA), and contains a chloramphenicol resistance gene. The Cas9 gene and the tracrRNA coding sequence are under the control of constitutive promoters while the λ Red gene cassette is transcribed using an arabinose-inducible promoter (pBAD). - The pCRISPR-SacB plasmid (Zerbini et al., 2017) is available from our laboratory upon request. It derives from the pCRISPR plasmid and the pKM154 plasmid from Addgene (#42875 and #13036, respectively).
Briefly, pCRISPR-SacB contains a cloning site, which allows insertion of a self-designed (see Procedure B), synthetic DNA fragment coding for the desired guide RNA, between two repeat strings under the constitutive PLtetO-1 promotor. It also carries a kanamycin resistance gene (KmR) fused to the sacB gene encoding the Bacillus subtilis levansucrase.
- The pCasRed plasmid (Zerbini et al., 2017) derives from the pCas9 plasmid from Addgene (#42876) and is available from our laboratory upon request.
- Selection of Cas9 target sites and construction of pCRISPR-SacB-gDNA plasmid
Note: Selection of Cas9 target sites is crucial to avoid off-target effects of the Cas9 endonuclease. Figure 3 illustrates how to identify target sites.- Identify a PAM sequence within the target gene (5’-NGG-3’)
Note: The PAM region should be close to the 5’-end as this protocol aims to achieve knockout through inactivation of the target gene. - Select the 30 base pairs (bp) upstream of the identified PAM sequence (protospacer) and BLAST (Altschul et al., 1990) them against the E. coli BL21(DE3) genome (GenBank accession number CP010816) (or any other strain you are going to use).
Note: Make sure that these 30 bp do not include any complementary nucleotides among the PAM-proximal 10-12 nucleotides, defined as the seed region.
Besides, to avoid off-target effects, the remaining PAM-distal part of the chosen sequence should share less than 15% homology with any other region of the remaining genome. - Order two complementary single-stranded synthetic oligonucleotides covering the protospacer minus the PAM region as described above, and five additional nucleotides to support the ligation during the subsequent cloning step (for example see Figure 3). The will code for the gene-specific guide RNA.
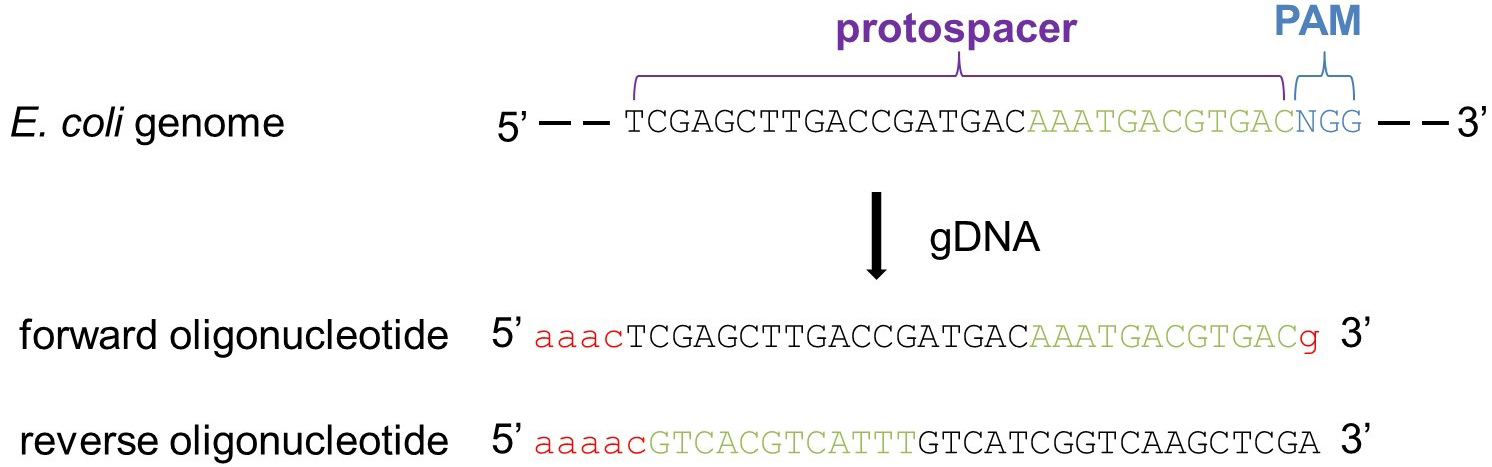
Figure 3. Illustration on how to select a gene-specific Cas9 target site. To design the guide DNA (gDNA) identify a PAM sequence (5’-NGG-3’, where N stands for any of the four nucleotides) near the 5’ end of the target gene. Then, select the 30 nucleotides upstream of the PAM region (protospacer). BLAST the entire sequence (protospacer + PAM), which is going to be the core of the forward oligonucleotide of your gDNA, against the genome of the target organism. To avoid Cas9 off-target effect, a ‘reliable’ gDNA should not have any homology among the 10-12 PAM-proximal nucleotides (seed region, in green) and less than 15% homology among the remaining protospacer when blasted against the rest of the genome. Finally, the chosen protospacer + PAM sequence is completed by the addition of five base pairs (in red) to facilitate the ligation for the subsequent cloning of the pCRISPR-SacB-gDNA, resulting in the forward oligonucleotide of the gDNA sequence. The reverse oligonucleotide of the gDNA is the reverse complement of the protospacer + PAM forward sequence with the addition of five base pairs (in red) as done for the forward oligonucleotide. - The synthetic DNA fragments have to be cloned into the pCRISPR-SacB as follows:
- Use T4 PNK to phosphorylate the two complementary single-stranded synthetic oligonucleotides encoding the gDNA.
1 µl oligonucleotide 1 (100 µM)
1 µl oligonucleotide 2 (100 µM)
5 µl 10x T4 ligase buffer
1 µl T4 PNK
42 µl MilliQ water (final volume 50 µl)
Note: If you have oligonucleotides diluted to concentrations other than 100 µM (e.g., 50 µM), make sure you adjust the volume of oligonucleotide 1 and 2 (e.g., 2 µl) and MilliQ water (e.g., 40 µl) to get a final volume of 50 µl. - Incubate for 30 min at 37 °C.
- For annealing, add 2.5 µl of 1 M NaCl to the reaction mix and heat to 95 °C for 5 min.
- Allow to cool down slowly to room temperature by simply switching off the thermoblock (ca. 2 h). In the meanwhile, proceed with Steps B4e-B4h.
- Digest the pCRISPR-SacB with BsaI.
10 µl pCRISPR-SacB (100 ng/µl, total amount ca. 1-2 µg)
1 µl BsaI
5 µl 10x NEB Buffer
34 µl MilliQ water (final volume 50 µl)
Note: If your pCRISPR-SacB plasmid is diluted to concentrations other than 100 ng/µl (e.g., 50 ng/µl), make sure you adjust the volume of the plasmid (e.g., 20 µl) and MilliQ water (e.g., 24 µl) to get a final volume of 50 µl. - Incubate for 1 h at 37 °C.
- Dephosphorylate the digested pCRISPR-SacB plasmid using Calf Intestinal Alkaline Phosphatase (CIP).
5 µl 10x CutSmart Buffer
1 µl CIP
25 µl pCRISPR-SacB plasmid cleaned in a PCR cleaning column (20 ng/µl, total amount of 500 ng plasmid)
19 µl MilliQ water
Note: If the purified pCRISPR-SacB plasmid is diluted to concentrations other than 20 ng/µl (e.g., 15 ng/µl), make sure you adjust the volume of the plasmid (e.g., 33.3 µl) and MilliQ water (e.g., 10.7 µl) to get a final volume of 50 µl. The concentration of your plasmid after purification should therefore not be lower than 11.4 ng/µl! - Incubate for 2 h at 37 °C.
- Purify the plasmid directly from the reaction mix using the Wizard® SV Gel and PCR Clean-Up System.
- To generate the pCRISPR-SacB-gDNA plasmid perform a final ligation step as follows:
3 µl digested pCRISPR-SacB (10 ng/µl, total amount of ca. 30 ng plasmid)
2 µl phosphorylated, double-stranded gDNA (1 μM)
1 µl T4 ligase
2 µl T4 ligase buffer
12 µl MilliQ water
Note: You may use different volumes, but make sure the concentrations remain the same in a final volume of 20 µl. - Incubate for 2 h at room temperature.
Note: Alternatively, if your time does not allow proceeding the same day, you may also incubate overnight at 16 °C. - The ligation reaction (10 µl) is used to transform E. coli DH5α chemically competent cells to obtain the pCRISPR-SacB-gDNA plasmid (see Step E2 for transformation via heat-shock).
Note: If you do not want to use commercial E. coli DH5α cells, you can also prepare your own batch of chemically competent cells (Step E1). - Incubate at 37 °C with vigorous shaking (200 rpm) for 1 h.
- Plate the transformation mix on LB-agar (see Recipes) containing kanamycin (50 µg/ml).
Note: As transformation efficiency should be high at this stage, do not plate the entire transformation mix but rather 100 µl or 200 µl only. - Incubate the LB-agar plate at 37 °C overnight.
- Inoculate 5 ml LB containing kanamycin (50 µg/ml) from a single colony and incubate at 37 °C overnight.
- Isolate the plasmid from the overnight culture using a QIAprep Spin Miniprep Kit.
- Analyze the plasmid by DNA sequencing using a primer (primer sequence: 5’-GCCCAGTCATAGCCGAATAG-3’) for the plasmid annealing downstream of the inserted fragment encoding the guide. The primer can be used for all pCRISPR-SacB-gDNAs and is therefore universal for every cloning of different guides.
If positive, your pCRISPR-SacB-gDNA plasmid is ready for the following mutagenesis step (Step E4a.ii).
- Use T4 PNK to phosphorylate the two complementary single-stranded synthetic oligonucleotides encoding the gDNA.
- Identify a PAM sequence within the target gene (5’-NGG-3’)
- Design of mutagenic donor DNA and primers for PCR screening
- We recommend designing your mutagenic donor DNA (dDNA) in a way that will result in the deletion of 30 nucleotides (∆30) within your target gene through the removal of the protospacer and the PAM region (to avoid post-mutation Cas9 cleavage). At the same time, an in-frame stop codon has to be inserted close to the 5’-end of the target gene, thereby inactivating it. Note that the trinucleotide encoding the stop codon has no complementary sequence on the target strand (Figure 4). In our hands, this strategy works well to achieve gene knockout. For a schematic example, refer to Figure 4. Note that the length of the deletion can be adapted to your needs. However, in case of longer deletions you may have to increase also the length of the dDNA.
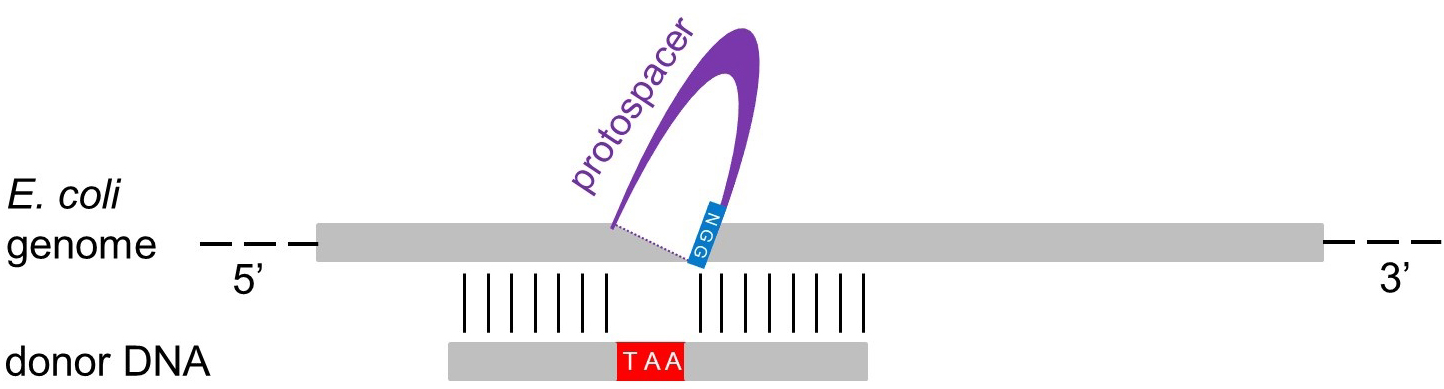
Figure 4. Design of the dDNA. The donor DNA (dDNA) can vary in length (e.g., 70 or 120 nucleotides, see text). To delete the protospacer + PAM region identified for Cas9 cleavage (Figure 3), the 5’ moiety of the dDNA (35 or 60 nucleotides) has to be complementary to the sequence upstream of the protospacer region, while the 3’ moiety of the dDNA (35 or 60 nucleotides) has to be complementary to the sequence downstream of the protospacer. To inactivate the transcript, an in-frame stop codon (TAA) is inserted, and the PAM region (NGG) is also eliminated to prevent further cleavage through Cas9. Finally, a reverse complement oligonucleotide is designed to get a double-stranded dDNA. - We explicitly recommend the use of double-stranded mutagenic oligonucleotides (ds-dDNAs), preferably in HPLC purification grade, as this considerably increases mutagenesis efficiency (Zerbini et al., 2017). Thus, design both forward and reverse oligonucleotides. For ∆30 deletions, we recommend dDNA of 70 nucleotides in length.
- Finally, the ds-dDNA is generated by annealing 10 µg of each complementary oligonucleotide pair in a total volume of 20 µl at 95 °C for 5 min and allowing the reaction mix to slowly cool down to room temperature by switching off the thermoblock. The annealing reaction is verified by loading 500 ng of each single stranded oligonucleotides and 1 µg of total DNA in the annealing reaction and by visualizing the bands using ATLAS ClearSight DNA Stain. The band corresponding to the single stranded oligonucleotides must disappear in the annealing reaction sample.
- The primers necessary for colony PCR screening following the mutagenesis step (Step E4a.ii) should be designed in order to get an amplified DNA fragment of ca. 200 bp, a fragment length that is ideal to distinguish the difference of a 30 bp deletion between the wild-type colony and a mutated colony. For a schematic example, refer to Figure 5.
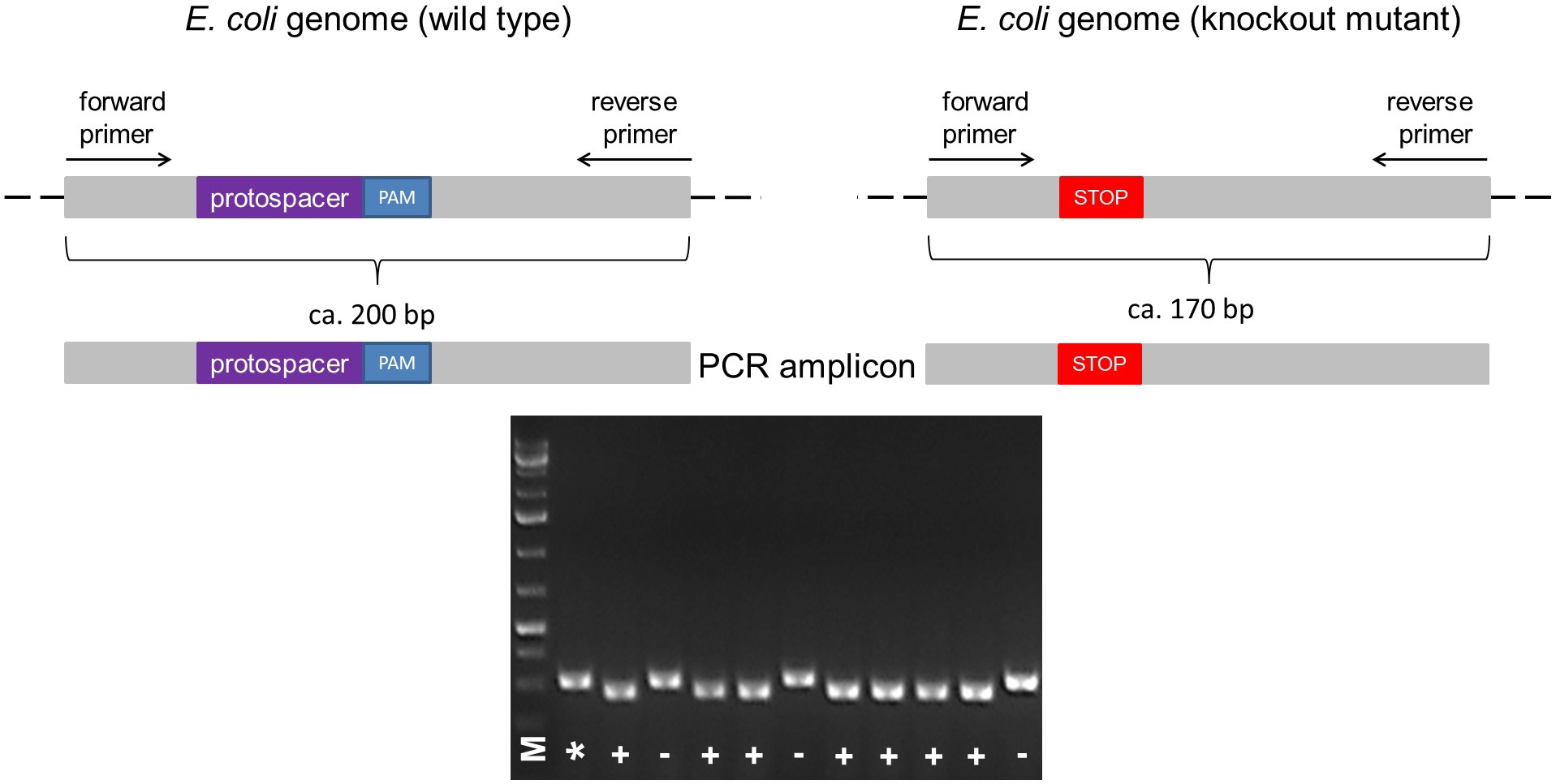
Figure 5. Illustration of the colony PCR. The forward and reverse primers for PCR are designed to anneal upstream and downstream of the protospacer + PAM sequence, respectively, resulting in the amplification of a ~200 bp fragment in the wild type. According to the size of deletion (e.g., Δ30), a knockout mutant can be identified by its shorter PCR fragment (e.g., ~170 bp) compared to the wild type (A). The PCR fragments different in size can be identified by running a 2% agarose gel electrophoresis (B). Legend: marker (M), wild type control (*), mutant (+), wild type ‘escaper’ (-).
- We recommend designing your mutagenic donor DNA (dDNA) in a way that will result in the deletion of 30 nucleotides (∆30) within your target gene through the removal of the protospacer and the PAM region (to avoid post-mutation Cas9 cleavage). At the same time, an in-frame stop codon has to be inserted close to the 5’-end of the target gene, thereby inactivating it. Note that the trinucleotide encoding the stop codon has no complementary sequence on the target strand (Figure 4). In our hands, this strategy works well to achieve gene knockout. For a schematic example, refer to Figure 4. Note that the length of the deletion can be adapted to your needs. However, in case of longer deletions you may have to increase also the length of the dDNA.
- Bacterial strains and culture conditions
Note: Make sure you use the correct antibiotic required for selection of your E. coli strain. We routinely use the DH5α and BL21(DE3) strain.- For any experimental step, grow the Escherichia coli strain in LB broth (Sigma-Aldrich) at 37 °C with vigorous shaking (200 rpm), if not stated otherwise.
- Prepare stocks of your strains in LB + glycerol (15-25% final concentration) and store them at -80 °C.
Example: For a stock of 1 ml prepare 200 μl of 75% glycerol and add 800 μl of your strain after overnight growth. - Start from an overnight culture taken from a frozen/glycerol stock before each bacterial manipulation step, if not stated otherwise.
- For any experimental step, grow the Escherichia coli strain in LB broth (Sigma-Aldrich) at 37 °C with vigorous shaking (200 rpm), if not stated otherwise.
- Gene knockout using CRISPR/Cas9
For preparation of the pCRISPR-SacB-gDNA plasmids (see Step B4) we recommend using E. coli DH5α cells, which have to be made chemically competent and transformed via heat-shock (Steps E1 and E2). Instead, the strain you want to use for all mutagenesis experiments should be prepared for electroporation (Steps E3 and E4) to increase transformation efficiency.
The strain should be first transformed via electroporation (Step E4) with the pCasRed plasmid (from Step A1) before transforming via electroporation with the pCRISPR-SacB-gDNA plasmid (generated in Step B4).- Preparation of chemically competent cells
- Streak E. coli DH5α on an LB-agar plate and incubate at 37 °C overnight.
- Inoculate a pre-culture of 5 ml LB medium (see Recipes) from a single colony and incubate at 37 °C with vigorous shaking (200 rpm) overnight.
- Dilute the overnight culture to an OD600 nm of 0.1 in LB (e.g., 50 ml) and incubate at 37 °C with vigorous shaking (200 rpm) until an OD600 nm of 0.4-0.6 is reached (ca. 2 h).
- Transfer the culture to a 50 ml tube and harvest the cells by centrifugation at 4 °C, 2,500 x g for 20 min.
- Discard the supernatant and resuspend the pellet in 5 ml of ice-cold, sterile 100 mM MgCl2.
- Keep the suspension on ice for 30 min.
- Harvest the cells by centrifugation at 2,500 x g for 20 min.
- Discard the supernatant and gently resuspend the cell pellet in 1 ml of ice-cold, sterile 100 mM CaCl2 in 15% glycerol and prepare aliquots of 110 µl.
Note: If you started with a higher culture volume keep a ratio of 10 ml 100 mM CaCl2 in 15% glycerol/50 ml starting culture for resuspension. - Use the aliquots freshly for the subsequent transformation via heat-shock (see Step E2)
Note: If some aliquots remain, store them at -80 °C.
- Streak E. coli DH5α on an LB-agar plate and incubate at 37 °C overnight.
- Transformation via heat-shock
- Mix 50 μl of chemically competent cells with 10 μl of ligation mix (see Step B4l).
- Keep the reaction mix on ice for 30 min.
- Transfer the cells to a thermoblock set at 42 °C for 45 sec.
- Keep the cells on ice for 2 min.
- Add 940 µl LB and incubate the cells at 37 °C with vigorous shaking (200 rpm) for 1 h.
- Plate the cells on LB-agar plates containing the appropriate antibiotic (chloramphenicol 25 µg/ml for transformation with pCasRed plasmid or kanamycin 50 µg/ml for transformation with pCRISPR-SacB-gDNA)
Note: We recommend plating 100 μl and 900 μl of the transformation mix separately, but if you prefer you may choose different volumes (e.g., 500 µl or 250 µl). - Incubate at 37 °C overnight.
- Count the colonies to determine the transformation efficiency of your chemically competent cells.
- Mix 50 μl of chemically competent cells with 10 μl of ligation mix (see Step B4l).
- Preparation of electrocompetent cells
- Streak your E. coli strain on an LB-agar plate and incubate at 37 °C overnight.
- Inoculate a pre-culture of 5 ml LB medium (see Recipes) from a single colony and incubate at 37 °C with vigorous shaking (200 rpm, overnight)
Note: If you already performed a mutation round, use the overnight culture from the plasmid curing step (see Step E7). - Dilute the overnight culture to an OD600 nm of 0.1 in LB (e.g., 50 ml) and incubate at 37 °C wih vigorous shaking (200 rpm) until an OD600 nm of 0.6-0.8 is reached (ca. 2-3 h).
Note: If you prepare competent cells from a strain previously transformed with the pCasRed plasmid, make sure your media contains chloramphenicol (25 µg/ml) and add L-(+)-arabinose (final concentration of 0.2%, see Recipes) at an OD600 nm of 0.2 to the cells for the induction of λ Red genes. - Transfer the culture into a 50 ml tube and harvest the cells by centrifugation at 4 °C, 4,100 x g for 20-30 min.
- Discard the supernatant and resuspend the pellet in 50 ml ice-cold, sterile MilliQ water. Centrifuge again at 4 °C, 4,100 x g for 10 min. Repeat this wash step three times.
- Finally, wash the pellet with ice-cold, sterile 10% glycerol and centrifuge again at 4 °C, 4,100 x g for 10 min.
- Discard as much supernatant as possible. Immediately resuspend the cell pellet in 250 µl ice-cold, sterile 10% glycerol and prepare 110 µl aliquots.
Note: If you started with a higher culture volume, keep a ratio of 250 µl 10% glycerol/50 ml starting culture for resuspension. - Use the aliquots freshly for the subsequent transformation via electroporation (see Step E4).
Note: If some aliquots remain, store them at -80 °C.
- Streak your E. coli strain on an LB-agar plate and incubate at 37 °C overnight.
- Transformation via electroporation
- Electroporate 50 μl of competent cells using a 1 mm Gene Pulser cuvette at 1.8 kV with one of the following reaction mixes:
1 ng of pCasRed plasmid (see Step A1)
Mutagenesis step:
100 ng pCRISPR-SacB-gDNA (prepared in Step B4)
10 μg of the corresponding ds-dDNA (designed in Procedure C).
Note: Your control should consist in a transformation using 100 ng of the ‘empty’ pCRISPR-SacB (see Step B2) without adding any ds-dDNA. - Add 950 µl LB and incubate the cells at 30 °C with vigorous shaking (200 rpm) for 3 h.
- Plate the cells on LB-agar plates containing chloramphenicol 25 µg/ml (and kanamycin 50 µg/ml, when performing the mutagenesis step).
Note: We recommend plating the entire transformation mix of the mutagenesis step (1 ml), but if you prefer you may choose different volumes (e.g., 500 µl or 250 µl). - Incubate at 37 °C overnight.
- Count the colonies to determine the transformation efficiency of your electrocompetent cells.
Note: The control of your mutagenesis step should count ca. 106 CFU/ml/μg. A drop in transformation efficiency of at least three orders of magnitudes is indicative for the quality of the gDNA (Zerbini et al., 2017).
- Electroporate 50 μl of competent cells using a 1 mm Gene Pulser cuvette at 1.8 kV with one of the following reaction mixes:
- Colony PCR
- Prepare a master mix for colony PCR using the GoTaq Green Master Mix.
Note: In our hands, screening of ten colonies is sufficient (Zerbini et al., 2017). Thus, prepare the master mix for 12 reactions (10 samples + 1 control, see Note from Step E4a.ii).
11 x 1 µl primer, forward (10 µM)
11 x 1 µl primer, reverse (10 µM)
11 x 12.5 µl GoTaq
11 x 10.5 µl MilliQ water - Pick individual colonies using a pipette tip and transfer directly to the PCR reaction mix. Make sure you will have enough cells from the colony for further analysis (see Step E7 and/or E8) by inoculating onto a transfer plate before dipping into the PCR reaction tube.
- Start PCR with the following settings:
- Initial denaturation 95 °C for 5 min
- 30 cycles of:
Denaturation 95 °C for 30 sec
Annealing 58-63 °C for 30 sec
Elongation 72 °C for 30 sec - Final elongation 72 °C for 3 min
Note: Annealing temperature depends on the primer sequence. We designed them to use a range between 58-63 °C.
- Prepare a master mix for colony PCR using the GoTaq Green Master Mix.
- Verify the mutants by loading 4-8 µl of each PCR on a 2% agarose gel and run electrophoresis. Purify positive PCR amplicons (Δ30 between control and sample) using the Wizard® SV Gel and PCR Clean-Up System. Confirm the mutation by DNA sequencing using one of the primers you used for colony PCR above.
- Curing of pCRISPR-SacB-gDNA plasmid
Note: If you are going to perform more than one knockout round, this step is mandatory. However, at the end of your genome editing experiment(s), removal of the pCRISPR-SacB-gDNA plasmid is optional.- To remove the pCRISPR-SacB-gDNA plasmid, pick a mutated colony (from Step E5b) and inoculate liquid LB medium containing 5% sucrose (see Recipes) and chloramphenicol (25 µg/ml).
- Incubate at 37 °C with vigorous shaking (200 rpm) overnight.
- Prepare a glycerol stock of your resultant strain for storage at -80 °C.
Note: If you wish to continue with a subsequent mutation round, use the overnight culture for preparation of competent cells and return to Step E3. - Control loss of the pCRISPR-SacB-gDNA plasmid by adding 10-20 µl drops on two LB-agar plates, one containing chloramphenicol (25 µg/ml) the other one containing chloramphenicol and kanamycin (25 µg/ml and 50 µg/ml, respectively). No growth should be visible on the plate with the Cm and Km combination, indicating that all bacteria had lost the pCRISPR-SacB-gDNA plasmid.
- To remove the pCRISPR-SacB-gDNA plasmid, pick a mutated colony (from Step E5b) and inoculate liquid LB medium containing 5% sucrose (see Recipes) and chloramphenicol (25 µg/ml).
- Curing of pCasRed plasmid
Note: This step is optional. It is required if the presence of the pCasRed plasmid after the conclusion of your knockout experiment(s) may affect downstream experiments negatively. However, after removal of the pCasRed plasmid you may have to add a selection marker (i.e., antibiotic resistance) of your choice.- To remove the pCasRed plasmid, pick a mutated colony (from Step E5b) or use a glycerol stock, inoculate in LB medium without any antibiotic, and incubate at 37 °C with vigorous shaking (200 rpm) overnight.
- Confirm the loss of the pCasRed plasmid by inoculating LB-agar plate containing chloramphenicol (25 µg/ml). No growth should be visible when using chloramphenicol, indicating that all bacteria have lost the pCasRed plasmid.
- To remove the pCasRed plasmid, pick a mutated colony (from Step E5b) or use a glycerol stock, inoculate in LB medium without any antibiotic, and incubate at 37 °C with vigorous shaking (200 rpm) overnight.
- Preparation of chemically competent cells
Data analysis
To verify if the mutation has been successfully obtained, a colony PCR must be performed (see Step E5) and subsequently analyzed via gel electrophoresis using a 2% agarose gel (see Step E6). The size shift can be observed and documented using a gel documentation system (Figure 5). If a colony results to have a fragment equal to the wild type control, it is a so-called escaper (Jiang et al., 2013).
Finally, DNA sequencing using one of the gene specific primers employed also in colony PCR (Step E5) must be performed to confirm the mutation.
Recipes
- LB medium (liquid)
- Weigh 25 g of LB broth Miller powder
- Add MilliQ water to a final volume of 1 L
- Stir until the solution is fully dissolved
- Autoclave the medium
- Store the solution at room temperature
- Weigh 25 g of LB broth Miller powder
- LB-agar (for Petri dishes)
- Weigh 20 g of LB agar powder
- Add MilliQ water to a final volume of 500 ml
- Stir until the solution is fully dissolved
- Autoclave the medium
Note: If you do not pour the plates immediately, store the medium at room temperature. - Allow the medium to cool down to ca. 45-50 °C before adding the antibiotics and mix
- Pour the medium (ca. 10-15 ml) in Petri dishes
- Store your LB agar plates at 4 °C
- Weigh 20 g of LB agar powder
- Antibiotics
Note: We recommend preparing aliquots of 1,000x stock solutions for each antibiotic. Store them at -20 °C.- Chloramphenicol: 25 mg/ml
- Weigh 250 mg of chloramphenicol powder under a safety hood
- Add MilliQ water to a final volume of 10 ml
- Mix until the solution is fully dissolved
- Sterilize the solution with a 0.22 µm syringe filter
- Prepare 1 ml aliquots and store at -20 °C
- Kanamycin: 50 mg/ml
- Weigh 500 mg of kanamycin powder under a safety hood
- Add MilliQ water to a final volume of 10 ml
- Mix until the solution is fully dissolved
- Sterilize the solution with a 0.22 µm syringe filter
- Prepare 1 ml aliquots and store at -20 °C
- 20% L-(+)-arabinose
- Weigh 10 g of L-(+) arabinose
- Add MilliQ water to a final volume of 50 ml
- Mix until the solution is fully dissolved
- Sterilize the solution with a 0.22 µm syringe filter
- Store at room temperature
- Weigh 10 g of L-(+) arabinose
- 50% sucrose
- Weigh 100 g of sucrose
- Add MilliQ water to a final volume of 200 ml
- Mix until the solution is fully dissolved
- Sterilize the solution with a Primo® Vacuum Filter System, 0.22 μm, PES, 250 ml
- Store the solution at RT
- Weigh 100 g of sucrose
- 75% glycerol solutions
- Take 75 ml of autoclaved 100% glycerol
- Add 25 ml of autoclaved MilliQ water
- Mix thoroughly
- Take 75 ml of autoclaved 100% glycerol
- 100 mM MgCl2
- Weigh 95.21 mg of MgCl2 and dissolve in 10 ml MilliQ water
- Mix thoroughly
- Sterilize the solution with a 0.22 µm syringe filter
- Weigh 95.21 mg of MgCl2 and dissolve in 10 ml MilliQ water
- 100 mM CaCl2 in 15% glycerol
- Weigh 147.01 mg of CaCl2 dihydrate and dissolve in 8.5 ml MilliQ water
- Add 1.5 ml of sterile 100% glycerol
- Mix thoroughly
- Sterilize the solution with a 0.22 µm syringe filter
- Weigh 147.01 mg of CaCl2 dihydrate and dissolve in 8.5 ml MilliQ water
Acknowledgments
We want to acknowledge the financial support for this project provided through the Advanced ERC Grant OMVac 340915 assigned to Guido Grandi. The funding allowed us to set-up this protocol, which has been previously published in Microbial Cell Factories (Zerbini et al., 2017).
Further, we are grateful to the plasmid depository Addgene, which provided access to the plasmids pCas9 and pCRISPR that we modified for our purposes. Finally yet importantly, we want to thank Renate Weizbauer for the kind invitation to publish our protocol here.
We declare to not have any conflict of interest nor competing interests related to this article.
References
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. and Lipman, D. J. (1990). Basic local alignment search tool. J Mol Biol 215(3): 403-10.
- Datta, S., Costantino, N. and Court, D. L. (2006). A set of recombineering plasmids for Gram-negative bacteria. Gene 379: 109-115.
- Ellis, H. M., Yu, D., DiTizio, T. And Court, D. L. (2001). High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. P Natl Acad Sci USA 98(12): 6742-6746.
- Gay, P., Le Coq, D., Steinmetz, M., Berkelman, T. and Hoch, J. A. (1985). Positive selection procedure for entrapment of insertion sequence elements in Gam-negative bacteria. J Bacteriol 164(2): 918-921.
- Gay, P., Le Coq, D., Steinmetz, M., Ferrari, E. and Hoch, J. A. (1983). Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J Bacteriol 153(3): 1424-1431.
- Hale, L., Lazos, O., Haines, A. and Thomas, C. (2010). An efficient stress-free strategy to displace stable bacterial plasmids. Biotechniques 48(3): 223-228.
- Iyama T. and Wilson III, D. M. (2012). DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 12(8): 620-636.
- Jiang, W., Bikard, D., Cox, D., Zhang, F. and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR Cas systems. Nat Biotechnol 31(3): 233-239.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816-821.
- Ledford, H. (2015). CRISPR, the disruptor. Nature 522: 20-24.
- Marraffini, L. A. (2015). CRISPR-Cas immunity in prokaryotes. Nature 526: 55-61.
- Murphy, K. C. (1998). Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol 180(8): 2063-2071.
- Murphy, K. C., Campellone, K. G. and Poteete, A. R. (2000). PCR-mediated gene replacement in Escherichia coli. Gene 246(1-2): 321-330.
- Muyrers, J. P., Zhang, Y., Testa, G. and Stewart, A. F. (1999). Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res 27(6): 1555-1557.
- Pyne, M. E., Moo-Young, M., Chung, D. A. and Chou, C. P. (2015). Coupling the CRISPR/Cas9 system with lambda red recombineering enables simplified chromosomal gene replacement in Escherichia coli. Appl Environ Microb 81(15): 5103-5114.
- Sander, J. D. and Joung, J. K. (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32(4): 347-355.
- Shuman, S. and Glickman, M. S. (2007). Bacterial DNA repair by non-homologous end joining. Nat Rev Microbiol 5(11): 852-861.
- Steinmetz, M., Le Coq, D., Djemia, H.B. and Gay, P. (1983). Genetic analysis of sacB, the structural gene of a secreted enzyme, levansucrase of Bacillus subtilis Marburg. Mol Gen Genet 191(1): 138-144.
- Wright, D. G., Castore, R., Shi, R., Mallick, A., Ennis, D. G. and Harrison, L. (2016). Mycobacterium tuberculosis and Mycobacterium marinum non-homologous end-joining proteins can function together to join DNA ends in Escherichia coli. Mutagenesis 32(2): 245-256.
- Zerbini, F., Zanella, I., Fraccascia, D., König, E., Irene, C., Frattini, L. F., Tomasi, M., Fantappiè, L., Ganfini, L., Caproni, E., Parri, M., Grandi, A., Grandi, G. (2017). Large scale validation of an efficient CRISPR/Cas‑based multi gene editing protocol in Escherichia coli. Microb Cell Fact 16: 68.
- Zhang, Y., Buchholz, F., Muyrers, J. P. and Stewart, A. F. (1998). A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20(2): 123-128.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
König, E., Zerbini, F., Zanella, I., Fraccascia, D. and Grandi, G. (2018). Multiple Stepwise Gene Knockout Using CRISPR/Cas9 in Escherichia coli. Bio-protocol 8(2): e2688. DOI: 10.21769/BioProtoc.2688.
Category
Microbiology > Microbial genetics > Mutagenesis
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.