- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Chromatin Affinity Purification (ChAP) from Arabidopsis thaliana Rosette Leaves Using in vivo Biotinylation System
Published: Vol 8, Iss 1, Jan 5, 2018 DOI: 10.21769/BioProtoc.2677 Views: 9635
Reviewed by: Amey G RedkarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Chromatin Affinity Purification (ChAP) is widely used to study chromatin architecture and protein complexes interacting with DNA. Here we present an efficient method for ChAP from Arabidopsis thaliana rosette leaves, in which in vivo biotinylation system is used. The chromatin is digested by Micrococcal Nuclease (MNase), hence the distribution of nucleosomes is also achieved. The in vivo biotinylation system was initially developed for Drosophila melanogaster (Mito et al., 2005), but the presented protocol has been developed specifically for Arabidopsis thaliana (Sura et al., 2017).
Keywords: Chromatin affinity purificationBackground
Chromatin Immunoprecipitation (ChIP) became one of the most important and commonly used technique to study chromatin structure and organization. However, it requires good-quality antibodies, which will not cross-react with non-specific targets. This is relatively difficult to achieve in plants, which contain cell wall and are rich in photosynthesis-related compounds and proteins frequently causing cross-reactivity problems. On the other hand, obtaining stable transgenic organisms is a routine and easy strategy in plants. For these reasons most plant researchers choose gene tagging, where fusion proteins are obtained and used to study chromatin in an approach alternative to ChIP, that is Chromatin Affinity Purification (ChAP). The ChAP technique has proven to be extremely effective in plant chromatin studies (Zentner and Henikoff, 2014). Moreover, it is usually cheaper than classical ChIP as it does not require generation of antibodies, and is often more effective than ChIP as tags are recognized with higher affinity than antibodies raised directly against proteins of interests. One disadvantage of ChAP is that it cannot be used to study post-translational histone modifications. In the presented protocol, proteins are tagged with a short, Biotin ligase recognition peptide (BLRP), which is in vivo biotinylated by Escherichia coli BirA biotin ligase (de Boer et al., 2003). Consequently, the tagged protein is purified using streptavidin-based purification systems (e.g., Dynabeads M-280 Streptavidin, Invitrogen). As streptavidin has an extraordinarily high affinity for biotin (dissociation constant on the order of 10-14 mol/L), the binding of biotin to streptavidin is one of the strongest non-covalent interactions known in nature (Green, 1975). Alternatively, the presented protocol can be successfully applied for ChAP of proteins labeled with other tags (e.g., MYC, GFP, HA, FLAG) if a suitable system for final purification is used.
Materials and Reagents
- Standard pipette tips
- 15 ml and 50 ml conical centrifuge tubes
- Nylon mesh (pore size 80 μm, Membrane Solutions, catalog number: MENY090080 )
- Paper towels
- Miracloth (Merck, Calbiochem, catalog number: 475855 )
- 1.5-2.0 ml microcentrifuge tubes
- 2 ml and 1.5 ml low retention tubes (Maxymum recovery microtubes; Corning, Axygen®, catalog numbers: MCT-200-L-C and MCT-150-L-C )
- Plant material: A. thaliana plants expressing E. coli BirA biotin ligase under control of a strong promoter (e.g., Arabidopsis thaliana Act8) with a gene of interest fused with Biotin ligase recognition peptide (BLRP) (de Boer et al., 2003; Mito et al., 2005)
Note: Plants could be at different developmental stages from one-week-old seedlings to adult, prior flowering. In general, the younger material, the higher efficiency of chromatin isolation. Moreover, younger plants provide higher homogeneity of the tissue used for chromatin extraction and lower variation between replicates. BirA biotin ligase could be expressed from different promoters, however, expression should be kept at high levels. Other possible promoters include Ubiquitin 10 promoter (UBQ10) and Cauliflower Mozaic Virus (CaMV) 35S promoter. Inducible systems e.g., dexamethasone (DEX)-inducible, can be potentially also used, however this was not tested in our laboratory. - 2 M glycine (made of Glycine, Sigma-Aldrich, catalog number: 33226 )
Note: This product has been discontinued. - Liquid nitrogen, ice
- 1 M CaCl2 (made of CaCl2, Sigma-Aldrich, catalog number: C5670 )
- Micrococcal nuclease (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EN0181 )
- 0.5 M EGTA (made of EGTA, BioShop, catalog number: EGT101 )
- 0.5 M EDTA (made of EDTA, BioShop, catalog number: EDT001 )
- RNase A (Sigma-Aldrich, catalog number: R4642 )
- Proteinase K (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EO0491 )
- 3 M sodium acetate pH 5.2 (made of CH3COONa, INC Biomedicals Inc.)
- Glycogen (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0561 )
- Ethanol (Avantor Performance Materials, catalog number: BA6420113 )
- Agarose (BioShop, catalog number: AGA001.1 )
- Gene Ruler 1 kb, GR 100 bp and GR 100 bp plus (Thermo Fisher Scientific, Thermo ScientificTM, catalog numbers: SM0311 , SM0241 and SM0321 , respectively)
- Dynabeads M-280 Streptavidin (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11206D )
- Chelex 100 (Bio-Rad Laboratories, catalog number: 1421253 )
- Tris (BioShop, catalog number: TRS001.5 )
- 1 M Tris-HCl pH 8 (made of Tris)
- 1 M Tris-HCl pH 6.8 (as above)
- Acetic acid (Avantor Performance Materials, catalog number: BA8760114 )
- Sucrose (Sigma-Aldrich, catalog number: S0389 )
- Phenylmethylsulfonyl fluoride (Sigma-Aldrich, catalog number: P7626 )
- Formaldehyde (Sigma-Aldrich, catalog number: F8775-500ML or BioShop, catalog number: FOR201 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266 )
- Triton X-100 (Carl Roth, catalog number: 3051.3 )
- β-Mercaptoethanol (BioShop, catalog number: MER002 )
- Spermine (Sigma-Aldrich, catalog number: 85590-5G )
- 5 M NaCl (made of NaCl, Avantor Performance Materials, catalog number: 794121116 )
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771 )
- Sodium bicarbonate (NaHCO3) (Sigma-Aldrich, catalog number: S5761 )
- Phenol (Sigma-Aldrich, catalog number: P4557 )
- Chloroform (Avantor Performance Materials, catalog number: BA6420113 )
- Isoamyl alcohol (Avantor Performance Materials, catalog number: 485560111 )
- Maxima Sybr Green/ROX qPCR Master Mix (2x) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K0223 )
- Protease inhibitor cocktail (Sigma-Aldrich, catalog number: P8465 )
- 1x TE buffer (see Recipes)
- 1x TAE buffer (see Recipes)
- Cross-linking buffer (see Recipes)
- Honda buffer (see Recipes)
- TNE buffer (see Recipes)
- Extraction buffer (see Recipes)
- Pellet extraction buffer (see Recipes)
- Phenol/Chloroform/Isoamyl alcohol for DNA extraction (see Recipes)
- ChIP Dilution Buffer (see Recipes)
- Binding/Washing Buffer (see Recipes)
Equipment
- Pipettes (Gilson, model: Pipetman® Neo or PZ HTL, model: Discovery Comfort )
- Fume hood
- Desiccator connected to the pump
- Mortars and pestles
- Small funnels
- Scale (KERN & SOHN, catalog number: EG 220-3NM )
- Rotator (Cole-Parmer, Stuart, model: SB3 )
- Centrifuges:
Thermo Fisher Scientific, Thermo ScientificTM, model: HeraeusTM MultifugeTM X1R, catalog number: 75004250
Thermo Fisher Scientific, Thermo ScientificTM, model: HeraeusTM BiofugeTM StratosTM Centrifuge, catalog number: 75005282
Beckman Coulter, model: Allegra® X-30R Centrifuge, catalog number: A99471
Eppendorf, model: MiniSpin® plus , catalog number: 0040262
Gilson, model: CmC Lab CAPSULEFUGE PMC-880 - Thermomixer (Eppendorf, model: Thermomixer comfort , catalog number: 5355)
- Magnetic separator (Thermo Fisher Scientific, model: DynaMagTM-2, catalog number: 12321D )
- See Saw Rocker (Cole-Parmer, Stuart, model: SSL4 )
- Thermoblock (Grant Instruments, model: QBA2 )
- Vortex (Scientific Industries, model: Vortex-Genie 2 , catalog number: SI-0256)
- Systems for DNA electrophoresis (Mini-Sub Cell and Wide Mini-Sub Cell GT Complete Systems, Bio-Rad Laboratories, catalog numbers: 1640300 and 1640301 , respectively)
- Real-Time PCR System (Thermo Fisher Scientific, Applied BiosystemsTM, model: 7900HT Fast Real-Time PCR System, catalog number: 4329001 )
Software
- 7900HT Fast Real-Time PCR System Software: SDSv2.4 (available on Thermo Scientific website)
Procedure
- Cross-linking
Cross-linking covalently preserves DNA-protein interactions for the further preparation steps. However, the excessive fixation may hinder later chromatin shearing and recognition of protein tags/epitopes. Use fresh formaldehyde, as its effective concentration drops over time, especially when exposed to air and light. Vacuum is used to increase tissue penetration. At the quenching step glycine becomes a substrate for the excess of formaldehyde.- Collect 4 g of Arabidopsis thaliana rosette leaves into a 50 ml conical tube. If plants are dehydrated as in case of drought-treated material, 2 g of leaves can be enough to obtain similar efficiency of chromatin extraction. Cover leaves with a small piece of nylon mesh and close the tube with a riddled screw-cap.
- Add 37 ml of cross-linking buffer (see Recipes) (under the fume hood) and apply vacuum for 10 min at room temperature for cross-linking to achieve the pressure of 50 mBar. The plant tissue should look translucent after cross-linking.
- Stop cross-linking by adding 2.5 ml of 2 M glycine (to final conc. of 100 mM) and applying the vacuum infiltration for additional 5 min.
- Collect 4 g of Arabidopsis thaliana rosette leaves into a 50 ml conical tube. If plants are dehydrated as in case of drought-treated material, 2 g of leaves can be enough to obtain similar efficiency of chromatin extraction. Cover leaves with a small piece of nylon mesh and close the tube with a riddled screw-cap.
- Chromatin isolation and MNase digestion
- Wash plant tissues five times in sterile deionized water. Remove the water as much as possible by blotting the tissues between paper towels. This is important as ice crystals may destroy nuclei that would reduce efficiency of chromatin isolation. Freeze the tissue quickly in liquid nitrogen.
- Grind thoroughly the tissue to a very fine powder using pre-chilled mortars and pestles and ensure that the samples do not thaw during grinding.
- Transfer the plant powder into 50 ml conical tubes pre-cooled with liquid nitrogen. At this point you can store the material (for limited time) at -80 °C.
- Resuspend each sample in 20 ml of ice-cold Honda buffer (see Recipes).
- Vortex briefly (avoid floating) to mix and incubate on ice for 30 min with mixing on the rotator to homogenize the samples.
- Filter the homogenate through two layers of Miracloth and squeeze the Miracloth.
- Transfer the filtrate to a new 50 ml conical tube, and spin at 2,000 x g, 4 °C in a swing-out rotor (Heraeus) for 15 min.
- Resuspend the pellet very gently by pipetting in 20 ml of Honda buffer.
- Spin the suspension at 2,000 x g at 4 °C for 15 min.
- Repeat resuspension and centrifugation as above. There should be soft white pellet of nuclei forming at the bottom of the tube. If the pellet is still green, additional washes may be needed.
- Resuspend the pellet in 20 ml of Honda buffer without spermine.
- Spin the suspension at 2,000 x g at 4 °C for 8 min.
- Resuspend the nuclei pellet in 1 ml of ice-cold TNE buffer (see Recipes) and divide it into two 1.5 ml microcentrifuge tubes.
- In the presence of 4 mM CaCl2 (add 2 μl 1 M CaCl2 to each tube) add an appropriate number of MNase units (this needs to be established experimentally, we usually add between 8-20 U) to each tube and immediately place the tubes in a thermomixer pre-warmed to 37 °C. Incubate the tubes for 20 min with vigorous shaking (1,400 rpm) to liberate nucleosomes.
- Stop the reaction by placing the tubes on ice and adding 25 μl 0.5 M EGTA (to a final concentration of 25 mM).
- Spin at 14,000 x g in a microcentrifuge for 5 min at 4 °C.
- Save supernatant for further steps and store at -178 °C (liquid nitrogen). You can also save the pellet to check the efficiency of chromatin isolation and MNase digestion (the optional step [Step C3]).
- Wash plant tissues five times in sterile deionized water. Remove the water as much as possible by blotting the tissues between paper towels. This is important as ice crystals may destroy nuclei that would reduce efficiency of chromatin isolation. Freeze the tissue quickly in liquid nitrogen.
- Detection of MNase digestion efficiency by DNA isolation (especially useful when optimizing MNase concentration)
- Reverse cross-linking and protein digestion
Prior to DNA isolation, the crosslinks between DNA and proteins need to be reversed.- Take 70 μl of chromatin isolate and add 400 μl of extraction buffer (see Recipes).
- Add 20 μl of 5 M NaCl and incubate at 65 °C for at least 4 h to overnight to reverse cross-linking.
- Add 10 μl of 0.5 M EDTA, 20 μl 1 M Tris-HCl pH 6.8, 1.7 μl RNase A (30 mg/ml) and incubate for 30 min at 37 °C.
- Add 1 μl Proteinase K (20 mg/ml) and incubate for 1.5 h at 45 °C to digest proteins.
- Take 70 μl of chromatin isolate and add 400 μl of extraction buffer (see Recipes).
- DNA precipitation
- Add an equal volume (500 μl) of phenol/chloroform/isoamyl alcohol and vortex briefly.
- Centrifuge at 14,000 x g for 5 min at RT and transfer the supernatant to a 2 ml Eppendorf tube.
- Add 1/10 volume of 3 M sodium acetate pH 5.2, 4 μl glycogen (20 mg/ml) and 2.5 volume of 100% EtOH, mix by inverting the tube several times and incubate at -80 °C for at least 40 min to precipitate the DNA.
- Centrifuge the sample at 14,000 x g for 15 min at 4 °C.
- Discard the supernatant, wash the pellet with 500 μl of cold 70% EtOH, centrifuge again for 5 min at 4 °C.
- Discard the supernatant and dry the pellet at RT. Dissolve the pellet in 20 μl and check on the 1.5% agarose gel (Figure 1). You should observe mainly mononucleosomal DNA fragments (of size ~150 bp). You can compare it to DNA isolated from the pellet after nuclei treatment with MNase (see optional step [Step C3] below).
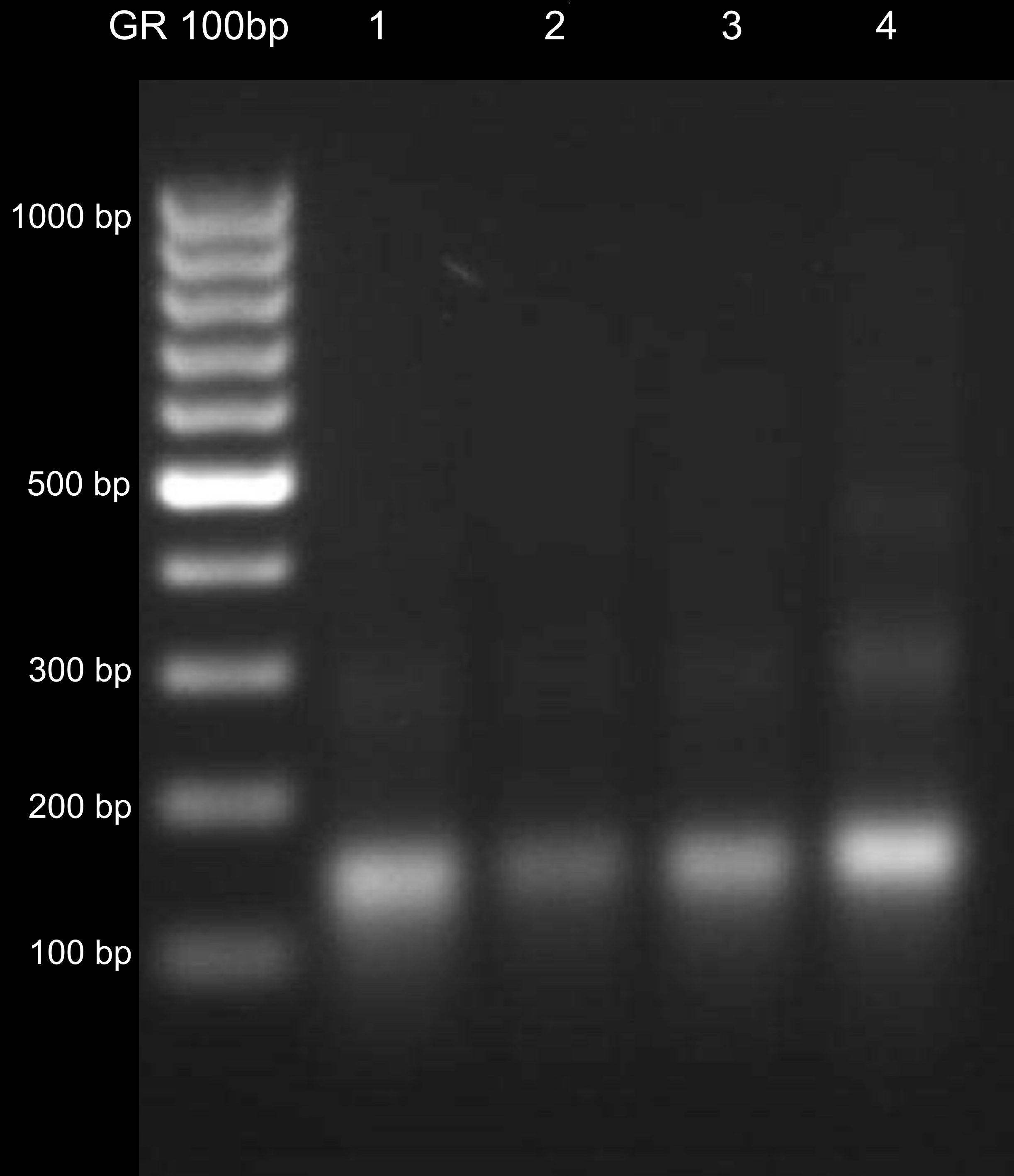
Figure 1. DNA isolated from MNase-treated chromatin. Lanes 1 to 4 correspond to different samples. DNA fragments of different length represent mono-, di- and trinucleosomal DNA (~150, ~300 and ~450 bp, respectively). The desired fragments are mononucleosomal, although overdigestion should be avoided as well.
- Add an equal volume (500 μl) of phenol/chloroform/isoamyl alcohol and vortex briefly.
- Optional: Isolation of DNA from pellet after MNase treatment
Note: This protocol can be used to estimate how much genomic DNA remained in the pellet.- Add 400 μl of Pellet extraction buffer (see Recipes) to the pellet obtained from MNase-treatment.
- Mix the tube vigorously for 15 min at 65 °C with 1,400 rpm shaking using thermomixer.
- Use 200 μl of the whole extract (no centrifugation) for incubation at 65 °C overnight to reverse crosslinking.
- Incubate with 1 μl RNase A (30 mg/ml) at 37 °C for 30 min.
- Add 1 μl of Proteinase K and incubate for 1.5 h at 45 °C.
- Add 200 μl phenol/chloroform/isoamyl alcohol (in a fume hood) to each sample and mix.
- Centrifuge at 16,000 x g for 5 min. Carefully transfer aqueous layer to fresh 1.5 ml tubes.
- Add 20 μl 3 M NaOAc to each tube, mix. Add 550 μl absolute ethanol, mix and incubate at -80 °C for at least 40 min.
- Centrifuge at 16,000 x g for 15 min at 4 °C to collect DNA.
- Wash pellet in 70% EtOH.
- Dry DNA pellet and resuspend in 50 μl TE buffer (see Recipes). Check 10 μl on the 1.5% gel along with a DNA ladder to estimate fragment size (Figure 2). You should observe that most of mono-, di-, and trinucleosomal DNA is present in the supernatant.
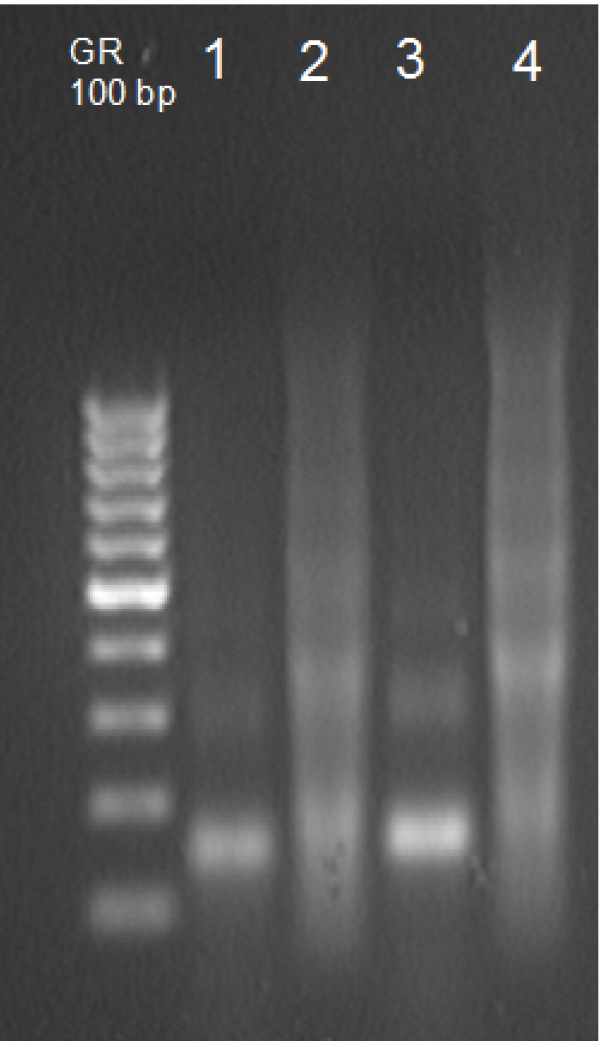
Figure 2. Verification of chromatin MNase digestion. Lanes 1 and 3 contain DNA isolated from the supernatant (control and stress plants, respectively), lanes 2 and 4 contain DNA from the pellet (control and stress plants, respectively).
- Add 400 μl of Pellet extraction buffer (see Recipes) to the pellet obtained from MNase-treatment.
- Reverse cross-linking and protein digestion
- Chromatin affinity purification (streptavidin to biotin)
- Take 25 µl of the Dynabeads M-280 Streptavidin slurry for each sample and put into the Maxymum Recovery (low retention) tube on ice.
- Wash three times each with 1 ml of ice-cold Binding/Washing Buffer (see Recipes). Collect the beads on tube wall on a magnetic separator (Figure 3).

Figure 3. Separation of streptavidin beads (Dynabeads M-280) using a magnetic separator. Beads attached to one side of Maxymum Recovery tube wall, so the supernatant can be easily removed, and the beads could be washed. - Resuspend the beads in 100 μl of cold Binding/Washing Buffer.
- Divide your chromatin supernatant (obtained in Step B17) into 140 μl portions (for ChAP) and 20 μl portions (for input).
- Dilute 140 μl of the chromatin supernatant ten times with ice-cold ChIP Dilution Buffer (add 1260 μl of ChIP Dilution Buffer) (with protease inhibitors added, see Recipes). Once you obtained a greater volume of the chromatin supernatant. Spin in a microcentrifuge at maximum speed for 10 min and then transfer to the tubes with beads (Step D3).
- Incubate overnight at 4 °C on a rotator such as Stuart SB3 Rotator with mild rotation (~15 rpm).
- Next morning, wash the beads on ice in the see-saw rocker:
5 x 5 min Binding/Washing Buffer
2 x 5 min TE - After final wash in TE, before beads collection, transfer the samples to fresh Maxymum Recovery tubes to avoid releasing DNA bound to the tube walls.
- Add 100 µl thoroughly mixed 10% Chelex (w/w) to the beads in each tube. Add 200 µl 10% Chelex to 20 µl input portion. Chelex is a chelating resin used to protect DNA from degradation at high temperatures (Singer-Sam et al., 1989) with proven efficacy during the elution step in ChIP method (Nelson et al., 2005). Exact volumes of chromatin used for ChAP or input and volumes of their eluates are needed in calculations of ‘percent of input’.
- Boil the samples in a heating block at 99 °C for 10 min.
- Add 1 µl Proteinase K (20 mg/ml) to each tube and incubate at 43 °C for 1 h.
- Inactivate the Proteinase K by incubation at 99 °C for 10 min.
- Spin the samples for 1 min at maximum speed and transfer the supernatant to new low retention tubes.
- Use 2 µl of ChAP and input eluates in qPCR reactions.
- Take 25 µl of the Dynabeads M-280 Streptavidin slurry for each sample and put into the Maxymum Recovery (low retention) tube on ice.
Data analysis
- In ChAP-qPCR at least three biological replicates should be used for each experiment (three ChAP replicates and three corresponding input replicates). Each biological replicate should be repeated in qPCR in at least three technical repeats and the mean Ct should be used for further calculations.
- As MNase digestion results in DNA fragments of about 150 bp length, primers should be designed to produce short amplicons (perfectly about 80 bp). The quality of primers is absolutely critical for the accuracy of calculations. They should result in a near-to-perfect amplification of DNA (the amount of DNA in each cycle should increase of > 1.9 times). This should be verified by running qPCR reactions using serial dilutions of template DNA of known concentration. Based on this amplification the standard curve should be calculated (Figure 4). The slope of the standard curve should not deviate significantly from -3.33 (perfect amplification) and the R2 of the standard curve should be > 0.985 (1.000 is a perfect amplification). To verify whether the amplified DNA is indeed the expected product (e.g., not primer-dimers), analysis of the dissociation curve is recommended.
- Together with ChAP samples, the corresponding input samples need to be analyzed. In this case, a dilution should be adjusted to a level in which both ChAP and input samples do not differ in their Ct values by more than 3 cycles.
- Having successfully verified raw results of the qPCR run, percent of input (% of input) may be calculated as follows (Lin et al., 2012):
% of input = 100 x 2ΔCt
where,
ΔCt = CtInput - log2(Input dilution factor) - CtChIP - To easily compare results from differentially treated plants or even different experiments, normalization to the reference region is commonly used. In principle, it is a region in a genome in which the occupancy and distribution pattern of our protein of interest does not change irrespective of used conditions/treatments.
Note: The example of such calculations is presented in the excel file.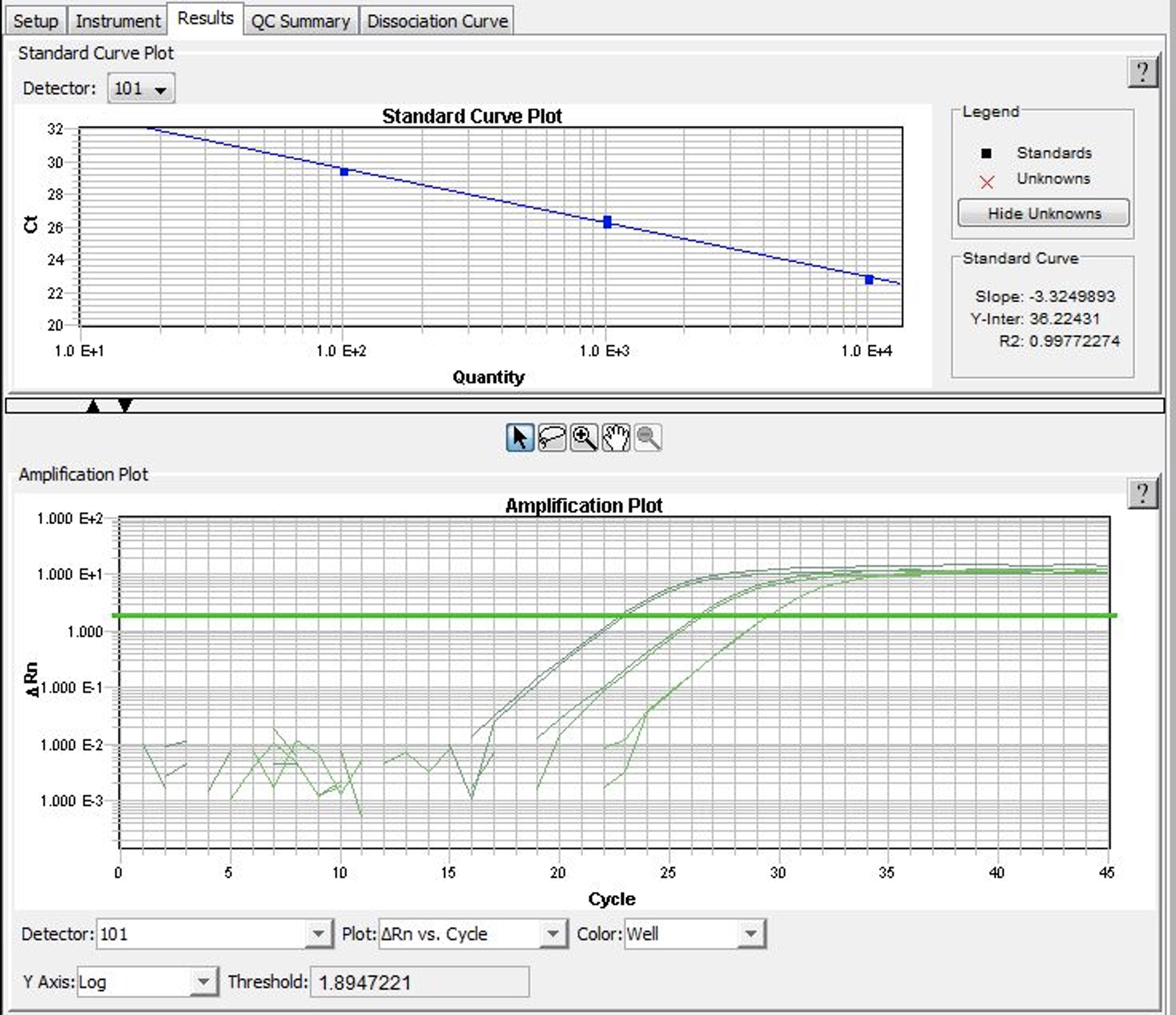
Figure 4. SDSv2.4 window printscreen. In the upper panel the standard curve plot calculated for a primer pair. The slope and R2 parameters inform about the PCR efficiency (ideal amplification has the slope close to -3.33 and R2 close to 1. In the bottom panel amplification plots for different DNA concentration. The software automatically sets the threshold and calculates Ct values.
Recipes
- 1x TE buffer
10 mM Tris-HCl (pH 8)
1 mM EDTA - 1x TAE buffer
40 mM Tris
20 mM acetic acid
1 mM EDTA - Cross-linking buffer
0.4 M sucrose
10 mM Tris-HCl (pH 8)
1 mM EDTA
1 mM PMSF
1% formaldehyde
Note: Make fresh buffer before each experiment. Presence of sucrose in the cross-linking buffer increases the efficiency of the DNA-protein cross-linking. The addition of the protease inhibitor (PMSF) should be done directly before using the buffer. - Honda buffer
25 mM Tris-HCl (pH 7.5)
0.44 M sucrose
10 mM MgCl2
0.5% Triton X-100
10 mM β-mercaptoethanol
2 mM spermine
Notes:- Sterilize by filtering (and store at 8 °C) or make fresh before use.
- Add spermine and β-mercaptoethanol (70 μl per 100 ml) just before use.
- The last washing is performed without spermine.
- Sterilize by filtering (and store at 8 °C) or make fresh before use.
- TNE buffer
10 mM Tris-HCl (pH 8.0)
100 mM NaCl
1 mM EDTA - Extraction buffer
0.625% SDS
0.125 M NaHCO3 - Pellet extraction buffer
1x TE pH 8.0
100 mM NaCl
2% Triton X-100
1% SDS - Phenol/Chloroform/Isoamyl alcohol for DNA extraction
25 parts of phenol (pH 8)
24 parts of chloroform
1 part of isoamyl alcohol
Note: Prepare fresh before use, mix vigorously before each pipetting. - ChIP Dilution Buffer
16.7 mM Tris-HCl (pH 8)
167 mM NaCl
1.2 mM EDTA
1.1% Triton X-100
1 mM PMSF*
Protease Inhibitor Cocktail* - Binding/Washing Buffer
20 mM Tris-HCl pH8
150 mM NaCl
2 mM EDTA
1% Triton X-100
0.1% SDS
1 mM PMSF*
*Note: Add directly just before use.
Acknowledgments
This protocol was developed from the previously published paper (Sura et al., 2017), and is partially based on protocols published elsewhere (Saleh et al., 2008; Wierzbicki et al., 2008; Zilberman et al., 2008). This work was supported by grants from the National Science Centre grants (2016/21/B/NZ2/01757 to P.A.Z. and 2015/17/N/NZ1/00028 to W.S.) and Polish Ministry of Science and Higher Education grants (N/N303/313437 to P.A.Z., DI/2011/028641 to W.S.). The authors have no conflict of interest or competing interests to declare.
References
- de Boer, E., Rodriguez, P., Bonte, E., Krijgsveld, J., Katsantoni, E., Heck, A., Grosveld, F. and Strouboulis, J. (2003). Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc Natl Acad Sci USA 100(13): 7480-5.
- Green, M. N. (1975). Avidin. Adv Protein Chem 29: 85-133.
- Lin, X., Tirichine, L. and Bowler, C. (2012). Protocol: Chromatin immunoprecipitation (ChIP) methodology to investigate histone modifications in two model diatom species. Plant Methods 8(1): 48.
- Mito, Y., Henikoff, J. G. and Henikoff, S. (2005). Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet 37(10): 1090-1097.
- Nelson, J. D., Denisenko, O., Sova, P. and Bomsztyk, K. (2006). Fast chromatin immunoprecipitation assay. Nucleic Acids Res 34: 1-7.
- Saleh, A., Alvarez-Venegas, R. and Avramova, Z. (2008). An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat Protoc 3(6): 1018-1025.
- Singer-Sam, J., Tanguay, R. L. and Rjggs, A. O. (1989). Use of Chelex to improve PCR signal from a small number of cells. Amplifications: A Forum for PCR Users: 11.
- Sura, W., Kabza, M., Karlowski, W. M., Bieluszewski, T., Kus-Slowinska, M., Paweloszek, L., Sadowski, J. and Ziolkowski, P. A. (2017). Dual role of the histone variant H2A.Z in transcriptional regulation of stress-response genes. Plant Cell 29(4): 791-807.
- Wierzbicki, A. T., Haag, J. R. and Pikaard, C. S. (2008). Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 135(4): 635-648.
- Zentner, G. E. and Henikoff, S. (2014). High-resolution digital profiling of the epigenome. Nat Rev Genet 15(12): 814-827.
- Zilberman, D., Coleman-Derr, D., Ballinger, T. and Henikoff, S. (2008). Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456 (7218): 125-129.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sura, W. and Ziolkowski, P. A. (2018). Chromatin Affinity Purification (ChAP) from Arabidopsis thaliana Rosette Leaves Using in vivo Biotinylation System. Bio-protocol 8(1): e2677. DOI: 10.21769/BioProtoc.2677.
Category
Plant Science > Plant molecular biology > Chromatin
Molecular Biology > DNA > DNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.