- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Rolling Circle Amplification to Screen Yam Germplasm for Badnavirus Infections and to Amplify and Characterise Novel Badnavirus Genomes
Published: Vol 8, Iss 1, Jan 5, 2018 DOI: 10.21769/BioProtoc.2672 Views: 10529
Reviewed by: Modesto Redrejo-RodriguezAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Since the first discovery of badnaviruses (family Caulimoviridae, genus Badnavirus) in yam (Dioscorea spp.) germplasm in the 1970s (Harrison and Roberts, 1973), several hundred partial badnavirus reverse transcriptase (RT)-ribonuclease H (RNaseH) sequences have been characterised (Kenyon et al., 2008; Bousalem et al., 2009), but only a few complete Dioscorea bacilliform virus (DBV) genome sequences have been reported (Phillips et al., 1999; Seal and Muller, 2007; Bömer et al., 2016 and 2017; Sukal et al., 2017; Umber et al., 2017). We have optimised a workflow involving total nucleic acid extractions and rolling circle amplification (RCA) combined with restriction enzyme analysis for the detection and amplification of DBVs present in yam germplasm. We have employed this approach successfully revealing three novel episomal yam badnaviruses (Bömer et al., 2016). We proposed this to be a complementary method to denaturing gradient gel electrophoresis, which enables a rapid indication of badnavirus diversity as well as the identification of potentially integrated badnavirus sequences in the host genome (Turaki et al., 2017). Here, we describe the step-by-step protocol to screen yam germplasm for badnavirus infections using RCA as an efficient research tool in the amplification and characterization of novel badnavirus genomes.
Keywords: YamBackground
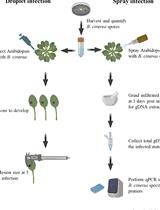
RCA is a sequence-independent strategy regularly used for the amplification of circular DNA virus genomes (Rector et al., 2004). Phi29 polymerase-mediated RCA techniques for (i) the detection of novel viruses; (ii) whole genome analysis of viruses; (iii) the generation of infectious clones; (iv) virus diagnostics of known viruses; (v) identification of replicative forms of viral genomes and; (vi) the differentiation between linear and circular forms are important tools in virology (reviewed by Johne et al., 2009). Circular target sequences are amplified more efficiently in RCA reactions as compared to linear templates. RCA has been recognized as a method of choice for the detection of Banana streak virus, also enabling the discrimination between integrated and episomal virus sequences (James et al., 2011) and was proposed to be used for DBV diagnostic purposes in yam (Umber et al., 2014). We uncovered several limitations to the usefulness of RCA in DBV diagnostics discussed in our previous study, including the amplification of putatively integrated sequences at lower frequencies and the amplification of circular plant plastid sequences. A relatively high chance for false-negative results also exists due to potential inhibition of the RCA assay due to plant compounds present in yam DNA extractions. We concluded that RCA is a useful tool for DBV research purposes (Figure 1, Bömer et al., 2016), which has been demonstrated several times since then (Bömer et al., 2017; Sukal et al., 2017). This led to the amplification and characterisation of several new full-length yam badnavirus genomes.
Figure 1. Scheme of the rolling circle amplification (RCA) method described in this protocol for the selective detection of episomal badnavirus genomes. RCA is a sequence-independent multiply-primed technique which can be used for the amplification and characterisation of episomal badnavirus sequences and was evaluated in yam germplasm in a previous study (Bömer et al., 2016).
Materials and Reagents
Note: The protocol described here is based on the successful RCA of three yam badnaviruses (Bömer et al., 2016).
- Consumables
- Gauge bags for sampling and grinding (10 x 15 cm) (Polybags, catalog number: 46500 )
- 1.5 ml microcentrifuge tubes (Alpha Laboratories, catalog number: LW2375 )
- 200 µl PCR tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: AB0266 )
- Pipette tips
- Petri dishes, round, 9 x 1.6 cm (Alpha Laboratories, catalog number: RC2260 )
- Polyvinylpyrrolidone (PVP) (MW 40 kDa) (Sigma-Aldrich, catalog number: PVP40T )
- QIAGEN Genomic-tip 20 G or 100 G columns (QIAGEN, catalog number: 10223 or 10243 )
- Plant material
Yam plants (e.g., Dioscorea rotundata and D. alata breeding lines and landraces maintained at the International Institute of Tropical Agriculture (IITA), Ibadan, Nigeria). Accession numbers are provided in the legends of Figures 4 and 5 - Competent cells and plasmids
- Reagents
- Deionized water (sterile)
- Nuclease-free, sterile deionized distilled water, e.g., HyCloneTM Water, Molecular Biology Grade (GE Healthcare, HyCloneTM, catalog number: SH30538.02 )
- Cloning
- Oligonucleotides (5’-3’), 10 µM
- Badna-FP_ATGCCITTYGGIITIAARAAYGCICC
- Badna-RP_CCAYTTRCAIACISCICCCCAICC
- M13F_TGTAAAACGACGGCCAGT
- M13R_CAGGAAACAGCTATGACC
- SP6_ATTTAGGTGACACTATAG
- T7F_TAATACGACTCACTATAGGG
- Badna-FP_ATGCCITTYGGIITIAARAAYGCICC
- Enzymes and buffers
Badna-PCR and colony-PCR- DreamTaq Green DNA polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0713 )
- 10x DreamTaq Green buffer (Thermo Fisher Scientific, Thermo ScientificTM, supplied with the enzyme)
- dNTP set (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0182 )
- Agarose (Bioline, catalog number: BIO-41025 )
- SYBRTM SafeTM DNA gel stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S33102 )
- DNA-loading dye, e.g., 6x gel loading dye, purple (New England Biolabs, catalog number: B7024 )
- DNA-size standard, e.g., GeneRulerTM 1 kb Plus DNA ladder (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SM1331 )
- Tris-Borate-EDTA (TBE) buffer (Sigma-Aldrich, catalog number: T4415 )
RCA
IllustraTM TempliPhi 100 Amplification Kit (Amersham Biosciences, catalog number: 25640010 )
Restriction enzyme digestion- HindIII (New England Biolabs, catalog number: R0104S )
- NEBufferTM 2.1 (New England Biolabs, catalog number: B7202S , supplied with the HindIII enzyme)
- PstI (New England Biolabs, catalog number: R0140S )
- NEBufferTM 3.1 (New England Biolabs, catalog number: B7203S , supplied with the PstI enzyme)
Ligation - DreamTaq Green DNA polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0713 )
- Antibiotics and blue:white selection of transformants
- Ampicillin (Sigma-Aldrich, catalog number: A9518 , stock 50 mg/ml in deionized water, filter sterilized)
- Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich, catalog number: I6758 , stock 0.1 M in deionized water, filter sterilized)
- X-Gal (5-Bromo-4-chloro-3-indolyl-β-D-galactopyranoside) (Sigma-Aldrich, catalog number: B9146 , stock 50 mg/ml in dimethylformamide)
- Ampicillin (Sigma-Aldrich, catalog number: A9518 , stock 50 mg/ml in deionized water, filter sterilized)
- DNA purification
- PCR-purification, e.g., GeneJET PCR Purification Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K0701 )
- Gel-purification, e.g., reSourceTM Gel Purification Kit (Source BioScience, catalog number: SBS28706 )
- Plasmid isolation, e.g., GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K0502 )
- PCR-purification, e.g., GeneJET PCR Purification Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K0701 )
- Phenol:chloroform:isoamyl alcohol (25:24:1), saturated with 10 mM Tris, pH 8.0, 1 mM EDTA (Sigma-Aldrich, catalog number: P3803 )
- Chloroform:isoamyl alcohol (24:1) (Sigma-Aldrich, catalog number: C0549 )
- Isopropanol (Molecular Biology Grade, Fisher BioReagents) (Fisher Scientific, catalog number: BP2618212 )
- Ribonuclease A from bovine pancreas (RNase A) (Sigma-Aldrich, catalog number: R4642 )
- Liquid nitrogen
- Chemicals for CTAB nucleic acid extractions (see Recipes)
- CTAB solution
Cetyltrimethylammonium bromide (CTAB) (Sigma-Aldrich, catalog number: H6269 ) - Tris-HCl solution, pH 8
Tris base (Trizma® base, Sigma-Aldrich, catalog number: T6066 )
37% HCl (Sigma-Aldrich, catalog number: 258148 ) - EDTA solution, pH 8
Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E5134 )
Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 1064621000 ) - NaCl solution
Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653 )
Sodium sulfite (Sigma-Aldrich, catalog number: 71988 )
- CTAB solution
- LB medium for E. coli (see Recipes)
- Deionized water (sterile)
Equipment
Note: No specific equipment is required. Any appropriate device can be used.
- Balance
- Pipettes
- Homogenizer hand model (Bioreba, catalog number: 400010 )
- Water bath
- Fume hood
- SpeedVac Concentrator (e.g., Thermo Fisher Scientific, Thermo ScientificTM, model: SavantTM DNA 120 SpeedVacTM Concentrator )
- Thermocycler (e.g., Thermo Fisher Scientific, Applied BiosystemsTM, model: VeritiTM 96-Well Thermal Cycler )
- Thermoblock (e.g., Eppendorf, model: ThermoMixer® C )
- Plate incubator (37 °C)
- Shaker (37 °C)
- Horizontal gel-electrophoresis system (e.g., Fisher Scientific, model: FisherbrandTM sub-gel midi-plus )
- UV transilluminator (e.g., Syngene, model: G:BOX Chemi HR16 )
Software
- NEBcutter V2.0 (http://nc2.neb.com/NEBcutter2/)
- MEGA version 7.0 (http://www.megasoftware.net/)
- SnapGene® Viewer version 4.0.4 (from GSL Biotech; available at snapgene.com)
Procedure
- Total nucleic acid extraction from yam leaves modified from Lodhi et al. (1994).
Note: Work in a fume hood for most steps (at least Steps 1j-1l).- Prepare CTAB extraction buffer (Recipe 5) and preheat buffer to 60 °C in a water bath.
- Select individual leaf samples and collect fresh leaves in small gauge bags (10 x 15 cm). Process leaves immediately (Figures 2A and 2B).
- Weigh approximately 300 mg of leaf sample and place into a fresh small gauge bag (Figure 2C).
- Briefly snap freeze leaf material by dipping the gauge bag into liquid nitrogen. No liquid nitrogen should enter the bag (Figure 2D).
- Immediately grind frozen leaf tissue manually into a fine powder using homogenizer (Figure 2E).
- Grind until thawing begins and tissue forms a ‘smooth paste’. Add 1 ml preheated CTAB extraction buffer and briefly grind again using homogenizer (Figures 2F and 2G).

Figure 2. Sampling and grinding of yam leaves for total nucleic acid extractions. A. Yam leaf showing viral symptoms; B. Collecting yam leaf in small gauge bag; C. Weighing ~300 mg of fresh yam leaf material; D. Snap freezing leaf material in liquid nitrogen; E. Grinding frozen leaf tissue into fine powder using homogenizer; F. Grinding until thawing begins and tissue forms a ‘smooth paste’; G. Further grinding after the addition of CTAB extraction buffer. - Add a further 2 ml CTAB extraction buffer (final volume 1 ml per 100 mg fresh weight leaf material) and mix thoroughly using homogenizer.
- Aliquot 700 µl of sap each into three fresh 1.5 ml microcentrifuge tubes using wide bore or cut 1 ml pipette tips (Figures 3A and 3B). Keep on ice until all samples are processed.
- Briefly vortex samples and incubate at 60 °C for 30 min in a water bath. Occasionally mix samples by inverting the microcentrifuge tubes 20 times (approximately every 10 min) (Figure 3C).
- Allow samples to reach room temperature and add an equal volume (700 µl) of phenol:chloroform:isoamyl alcohol (25:24:1) (Figure 3D). (Optional: Chloroform:isoamyl alcohol (24:1) can be used instead.) Mix to emulsion by inverting the tubes 50 times and centrifuge at 15,000 x g for 10 min (Figure 3E).
- Carefully transfer the upper aqueous phase (~600 µl) (Figure 3F) into a fresh 1.5 ml microcentrifuge tube without disturbing the interphase and add an equal amount of chloroform:isoamyl alcohol (24:1), mix and spin as before.

Figure 3. Sampling and grinding of yam leaves for total nucleic acid extractions. A-B. Aliquot 700 µl of sap each into three fresh 1.5 ml microcentrifuge tubes using wide bore or cut 1 ml pipette tips. C. Incubate samples at 60 °C for 30 min in a water bath and mix samples by inverting the microcentrifuge tubes. D. Allow samples to reach room temperature and add an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1). E. Mix to emulsion by inverting the tubes. F. After centrifugation, carefully transfer the upper aqueous phase without disturbing the interphase. - Carefully transfer the upper aqueous phase (~400 µl) into a fresh 1.5 ml microcentrifuge tube taking extra care not to disturb the interphase.
- Precipitate DNA by adding 75 µl of 5 M NaCl (Recipe 4) and 450 µl of ice-cold isopropanol. Mix samples well and incubate at -20 °C for 1 h. Work on ice from here on.
- Centrifuge samples at 15,000 x g and 4 °C for 10 min and discard the supernatant.
- Wash the pellet twice in 500 µl of 70% ethanol with centrifugations at 15,000 x g and 4 °C for 5 min.
- Remove the 70% ethanol by decanting and air dry the pellet using a vacuum concentrator. Do not dry completely as the pellet will become difficult to resuspend.
- Resuspend pellet in 100 µl nuclease-free, sterile distilled deionized water (SDW) at 4 °C overnight.
Optional: For optimal RCA detection, DNA pellets can be resuspended in 2 ml resuspension buffer (Recipe 6) instead and combined (three aliquots from the beginning), followed by purification through QIAGEN Genomic-tip 20 G or 100 G columns according to the manufacturer’s instructions. Inclusion of this column purification step was found to increase the efficiency of RCA. The final pellets were resuspended in 200 µl nuclease-free SDW at 4 °C overnight.
- Prepare CTAB extraction buffer (Recipe 5) and preheat buffer to 60 °C in a water bath.
- Screen the yam DNA samples for badnavirus sequences by PCR using the generic badnavirus primer pair Badna-FP/-RP (Yang et al., 2003).
- Set up a 20 µl PCR reaction per extracted DNA sample as follows. Final concentrations are indicated in brackets:
2 µl 10x DreamTaq Green buffer (1x)
0.5 µl dNTPs (0.25 mM)
1 µl Badna-FP (0.5 µM)
1 µl Badna-RP (0.5 µM)
0.2 µl DreamTaq DNA polymerase (0.05 U/ µl)
1 µl template DNA (1 ng/ µl)
14.3 µl deionized water - The cycle conditions for PCR amplification are 95 °C for 5 min (initial denaturation), followed by 30 cycles of 94 °C for 20 sec (denaturation), 56 °C for 30 sec (annealing), 72 °C for 30 sec (elongation) and a final extension of 72 °C for 7 min.
- Load 5-20 µl of the PCR reaction on a 1.5% (w/v) agarose gel including 1x SYBR Safe DNA gel stain in 0.5x TBE buffer. The expected amplicon is 579 bp (Figure 4).
Optional: To confirm the suitability of DNA for PCR amplification, all DNA samples can be first screened using primers targeting the yam actin gene, as described by Silva et al. (2015).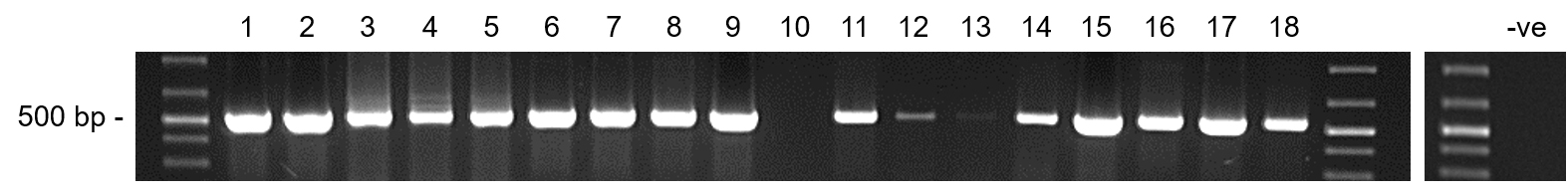
Figure 4. Screening yam DNA samples for badnavirus sequences by PCR. Typical agarose gel showing the expected amplicon size of 579 bp for Badna-PCR positive DNA samples from yam. Lane 1 = TDr 89/02475-A, Lane 2 = TDr 89/02475-B, Lane 3 = TDr 89/02677, Lane 4 = TDr 95/18544, Lane 5 = TDr 96/00629, Lane 6 = TDr 1892-A, Lane 7 = TDr 1892-B, Lane 8 = Pona, Lane 9 = TDr 00/00362-B, Lane 10 = TDr 94/01108-B, Lane 11 = Adaka-B, Lane 12 = TDr 03/00196-A, Lane 13 = TDr 00/00403, Lane 14 = Adaka-B, Lane 15 = TDr 00/00362-B, Lane 16 = TDr 00/00362-C, Lane 17 = TDr 94/01108-B, Lane 18 = TDr 03/00196-B. Adaka and Pona are yam landraces and yam accession numbers followed by A-C depicts clones of the same yam lines. -ve = PCR negative control which was included on a second gel.
- Set up a 20 µl PCR reaction per extracted DNA sample as follows. Final concentrations are indicated in brackets:
- Screen Badna-PCR positive yam DNA samples by RCA using IllustraTM TempliPhi 100 Amplification Kit.
- Set up an RCA reaction (final volume 10 µl) in PCR reaction tubes as follows. Final concentrations are indicated in brackets:
3.5 µl Kit sample buffer
0.25 µl Badna-FP (0.5 µM)
0.25 µl Badna-RP (0.5 µM)
1 µl undiluted DNA - Denature sample at 95 °C for 3 min (e.g., in a thermocycler) and chill immediately on ice.
- Prepare a master mix as follows:
5 µl Kit reaction buffer (per reaction)
0.2 µl Kit enzyme mix containing Phi29 polymerase (per reaction) - Incubate RCA reactions at 30 °C for 18 h using a thermocycler.
- Inactivate RCA reactions at 65 °C for 10 min.
Note: Do not freeze DNA samples before using them as templates in the RCA reactions but keep at 4 °C instead. This improves RCA reaction efficiencies.
- Set up an RCA reaction (final volume 10 µl) in PCR reaction tubes as follows. Final concentrations are indicated in brackets:
- Digest RCA products with restriction enzymes identified in silico (ideally single cutters, for example using software NEBcutter V2.0 [Vincze et al., 2003]) based on the viral genome sequence expected, or use for example HindIII or PstI when the viral genome sequence is unknown to generate useful restriction profiles.
- Digest 2 µl of RCA product according to the manufacturer’s instructions.
- Mix the digest with DNA-loading dye and load it on a 0.6% (w/v) agarose gel. Separate digested RCA restriction fragments by gel electrophoresis (Figure 5).
- Cut and purify RCA restriction fragments of interest from the gel. Use minimal UV exposure when cutting bands of interest. This will improve downstream cloning efficiency.
Note: A single cutter will produce an RCA restriction fragment of around 7.5 kbp, the typical size of a badnavirus genome. Mixed infections with unknown badnaviruses digested with HindIII and PstI will potentially produce very complex restriction patterns. See Figure 1 within Bömer et al. (2016) as an example of this.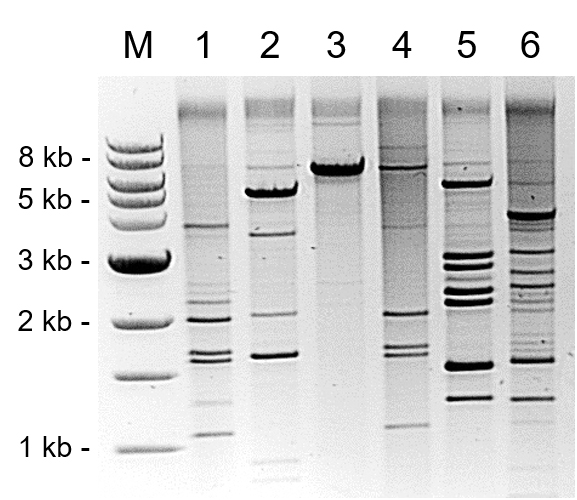
Figure 5. Separation of PstI-digested RCA restriction fragments from six Dioscorea species by gel electrophoresis. Restriction digestion patterns suggest the presence of different episomal badnavirus sequences as well as mixed infections in Dioscorea alata (TDa) and Dioscorea rotundata (TDr) accessions (Bömer et al., 2016). Some RCA products appear to be only partially digested. M = 1 kb DNA ladder, Lane 1 = TDr 89/02475, Lane 2 = TDa 00/00005, Lane 3 = TDa 85/00250, Lane 4 = TDr 95/18544, Lane 5 = TDa 95/00310, Lane 6 = TDr 1892.
- Digest 2 µl of RCA product according to the manufacturer’s instructions.
- (Optional) Screen purified RCA restriction fragments by PCR using the generic badnavirus primer pair Badna-FP/-RP (Yang et al., 2003). Sequence positive Badna-PCRs to assess the badnavirus diversity. This way, the focus can be channeled towards novel badnaviruses.
- Perform Badna-PCR reaction as described in Step 2 using a purified RCA restriction fragment as template.
- Purify positive Badna-PCRs and analyse the partial RT-RNaseH sequence by Sanger sequencing in both directions using the Badna-FP/-RP primer pair. The expected sequence is 579 bp long.
Optional: PCRs can be cloned before sequencing using the pGEM®-T Easy Cloning Kit according to the manufacturer’s instructions, or equivalent. The insert can then be sequenced with SP6 or T7F primer in one direction only.
- Perform Badna-PCR reaction as described in Step 2 using a purified RCA restriction fragment as template.
- Digest suitable cloning vector (e.g., pUC19 or pGEM®-3Zf (+)) with the same restriction enzyme chosen in Step 4 according to manufacturer’s instructions to linearize the vector.
- Mix the digest with DNA-loading dye and load it onto a 1% (w/v) agarose gel. Separate digested from undigested bands by gel electrophoresis.
Optional: Treat the linearized vector with alkaline phosphatase according to manufacturer’s instructions to prevent self-ligation. - Purify the linearized vector backbone (2.7 kbp for pUC19; 3.2 kbp for pGEM®-3Zf (+)) from the gel.
- Mix the digest with DNA-loading dye and load it onto a 1% (w/v) agarose gel. Separate digested from undigested bands by gel electrophoresis.
- Ligate the digested RCA fragment and the linearized vector.
- Set up a 10 µl ligation reaction in a 1.5 ml microcentrifuge tube as follows. Final concentrations are indicated in brackets:1 µl T4 DNA ligase 10x buffer (1x)
1 µl T4 DNA ligase (3 U)
Linearized vector (10 ng to 50 ng)
RCA restriction fragment (10 ng to 25 ng depending on size of insert and insert:vector ratio, e.g., 1:1 or 3:1)
Nuclease-free, sterile distilled deionized water ad 10 µl - Mix the reaction and incubate at room temperature for 1 h or at 4 °C overnight.
- Set up a 10 µl ligation reaction in a 1.5 ml microcentrifuge tube as follows. Final concentrations are indicated in brackets:1 µl T4 DNA ligase 10x buffer (1x)
- Use 2-4 µl of the ligation reaction to transform chemically competent NEB® 5-alpha E. coli (for pUC19) or JM109 E. coli (for pGEM®-3Zf (+)) cells according to the manufacturer’s instructions. Select transformants on LB plates (Recipe 7) with 100 µg/ml ampicillin, 0.5 mM IPTG and 80 µg/ml X-Gal (LB-amp, IPTG, X-Gal).
- Do colony-PCR to genotype transformants. Usually testing 5 to 10 colonies is sufficient to obtain positive transformants.
- Set up a 20 µl PCR reaction per colony as follows. Final concentrations are indicated in brackets:
2 µl 10x DreamTaq Green buffer (1x)
0.5 µl dNTPs (0.25 mM)
1 µl M13F (for pUC19; use SP6 for pGEM®-3Zf (+) instead) (0.5 µM)
1 µl M13R (for pUC19; use T7F for pGEM®-3Zf (+) instead) (0.5 µM)
0.2 µl DreamTaq DNA polymerase (1 U)
15.3 µl deionized water - Use a pipette tip to pick a white colony and restreak the cells on an LB-amp, IPTG, X-Gal plate. Afterwards, directly dip the tip into the PCR reaction to transfer a small amount of cells. Incubate the plate at 37 °C for approximately 5 h (or overnight) and run the PCR in the thermocycler.
Optional: The cells can be dipped into 20 µl sterile deionized water instead of the PCR reaction. The ‘colony-water’ is then incubated at 95 °C for 5 min and used as template (1 µl) for the colony-PCR. This approach allows repeating the PCR in case of failure. - The cycle conditions for PCR amplification were 95 °C for 5 min (initial denaturation), followed by 30 cycles of 94 °C for 30 sec (denaturation), 52 °C for 30 sec (annealing), 72 °C for 1 min/kbp insert size (elongation) and a final extension of 72 °C for 7 min.
- Load 5-20 µl of the PCR reaction on a 1.5% (w/v) agarose gel including 1x SYBR Safe DNA gel stain in 0.5x TBE buffer. The expected amplicon depends on the RCA restriction fragment size cloned.
- Prepare 5 ml of LB-amp and pick the PCR-positive clones from the restreak plate. Incubate overnight at 37 °C, 200 rpm.
- Isolate the plasmid from positive clones and analyse insert by Sanger sequencing using M13F/R or SP6/T7F primers to start with, as well as specific primers designed to walk the badnavirus genome.
Note: Colony-PCR will be inefficient if a complete badnavirus genome of around 7.5 kbp has been cloned due to the large size of the insert. Checking the presence and size of the insert can also be achieved by performing plasmid restriction digestion instead.
- Set up a 20 µl PCR reaction per colony as follows. Final concentrations are indicated in brackets:
Data analysis
Nucleotide sequences generated from plasmid clones and PCR products can be analysed and assembled using a sequence analysis tool, e.g., MEGA version 7.0 (http://www.megasoftware.net/). In all cases, a minimum of three independent clones should be sequenced in both directions to determine an accurate sequence of the putative full-length badnavirus genomes. Vector sequences can be removed using National Centre for Biotechnology Information (NCBI) VecScreen (http://www.ncbi.nlm.nih.gov/tools/vecscreen/) and the NCBI basic local alignment search tool (BLAST). Genome maps can be generated using SnapGene® Viewer version 4.0.4 (from GSL Biotech; available at snapgene.com). Edited sequences can be used for similarity BLAST searches in the NCBI GenBank databases (http://www.ncbi.nlm.nih.gov/genbank/). Multiple alignments of the partial RT-RNase H sequences can be performed using the CLUSTALW default settings in MEGA version 7, where phylogenetic relationships can also be analysed. This has been successfully performed in our previous study (Bömer et al., 2016).
Notes
RCA is widely used for the amplification of viral genomes and we recommend the review written by Johne et al. (2009). Reproducible DBV amplification in yam using RCA needs further improvements and is likely to be negatively affected by low viral titers of these badnaviruses as well as relatively high contents of inhibitory substances usually present in yam DNA extractions, such as phenolic compounds and polysaccharides. Immediate analysis of extracted DNA samples by RCA appears to make DBV amplification more efficient and is highly recommended. Thus, avoid unnecessary freezing cycles of extracted DNAs and store DNA extracts at 4 °C.
Recipes
- CTAB solution
10% (w/v) CTAB in deionized water
Autoclave CTAB solution at 121 °C and 2 bar for 15 min - Tris-HCl solution, pH 8 (1 L)
1 M Tris base
Adjust to pH 8 by adding concentrated HCl (~42 ml)
Autoclave Tris-HCl solution at 121 °C and 2 bar for 15 min - EDTA solution, pH 8 (1 L)
0.5 M EDTA
Adjust to pH 8 with NaOH (~20 g of NaOH pellets)
Autoclave EDTA solution at 121 °C and 2 bar for 15 min - NaCl solution
5 M NaCl in deionized water
Autoclave NaCl solution at 121 °C and 2 bar for 15 min - CTAB extraction buffer (10 ml)
2% (w/v) CTAB
0.1 M Tris-HCl, pH 8
0.02 M EDTA, pH 8
1.4 M NaCl
1% (w/v) sodium sulfite
2% PVP (MW 40 kDa)
Add deionized water to 10 ml
Optional: Add 1 µl of RNase A (20-40 mg/ml) per 1 ml CTAB extraction buffer for RNA removal
Note: Prepare CTAB extraction buffer from stock solutions immediately before use; the buffer is only good when freshly prepared. - Resuspension buffer (pH 7)
50 mM Tris-HCl
0.7 M NaCl
10 mM EDTA - LB medium for E. coli (1 L)
25 g of LB broth (Miller) for liquid LB or 35 g of LB Broth with agar (Lennox) for LB plates
1 L deionized water
Autoclave LB medium at 121 °C and 2 bar for 15 min
Note: Allow to cool slightly before making additions, such as antibiotics.
Acknowledgments
This protocol is based on our work previously published in Viruses (Bömer et al., 2016. Viruses 8: 188; doi: 10.3390/v8070188). The authors gratefully acknowledge the support of this work by the Bill & Melinda Gates Foundation (BMGF) under the ‘Development of On-farm Robust Diagnostic Toolkits for Yam Viruses’ grant to the Natural Resources Institute (NRI). Ajith Rathnayake was supported by a Vice-Chancellor PhD studentship awarded by the University of Greenwich. Research on yams at IITA was funded by the BMGF supported ‘Yam Improvement for Income and Food Security in West Africa (YIIFSWA)’ project and the CGIAR Research Program on Roots, Tubers and Bananas. The authors declare that they have no competing interests.
References
- Bömer, M., Rathnayake, A. I., Visendi, P., Silva, G. and Seal, S. E. (2017). Complete genome sequence of a new member of the genus Badnavirus, Dioscorea bacilliform RT virus 3, reveals the first evidence of recombination in yam badnaviruses. Arch Virol.
- Bömer, M., Turaki, A. A., Silva, G., Kumar, P. L. and Seal, S. E. (2016). A sequence-independent strategy for amplification and characterisation of episomal badnavirus sequences reveals three previously uncharacterised yam badnaviruses. Viruses 8(7).
- Bousalem, M., Durand, O., Scarcelli, N., Lebas, B. S., Kenyon, L., Marchand, J. L., Lefort, F. and Seal, S. E. (2009). Dilemmas caused by endogenous pararetroviruses regarding the taxonomy and diagnosis of yam (Dioscorea spp.) badnaviruses: analyses to support safe germplasm movement. Arch Virol 154(2): 297-314.
- Harrison, B. D. and Roberts, I. M. (1973). Association of virus-like particles with internal brown spot of yam (Dioscorea alata). Trop Agr (Trinidad) 50: 335-340.
- James, A. P., Geijskes, R.J., Dale, J. L. and Harding, R. M. (2011). Development of a novel rolling-circle amplification technique to detect Banana streak virus that also discriminates between integrated and episomal virus sequences. Plant Disease 95: 57-62.
- Johne, R., Muller, H., Rector, A., van Ranst, M. and Stevens, H. (2009). Rolling-circle amplification of viral DNA genomes using phi29 polymerase. Trends Microbiol 17(5): 205-211.
- Kenyon, L., Lebas, B. S. and Seal, S. E. (2008). Yams (Dioscorea spp.) from the South Pacific Islands contain many novel badnaviruses: implications for international movement of yam germplasm. Arch Virol 153(5): 877-889.
- Lodhi, M., Ye, G. N., Weeden, N. and Reisch, B. (1994). A simple and efficient method for DNA extraction from grapevine cultivars, Vitis species and Ampelopsis. Plant Molecular Biology Reporter 12: 6-13.
- Phillips, S., Briddon, R. W., Brunt, A. A. and Hull, R. (1999). The partial characterization of a badnavirus infecting the Greater Asiatic or water yam (Dioscorea alata). J Phytopathol 147: 265-269.
- Rector, A., Tachezy, R. and Van Ranst, M. (2004). A sequence-independent strategy for detection and cloning of circular DNA virus genomes by using multiply primed rolling-circle amplification. J Virol 78(10): 4993-4998.
- Seal, S. and Muller, E. (2007). Molecular analysis of a full-length sequence of a new yam badnavirus from Dioscorea sansibarensis. Arch Virol 152(4): 819-825.
- Silva, G., Bömer, M., Nkere, C., Kumar, P. L. and Seal, S. E. (2015). Rapid and specific detection of Yam mosaic virus by reverse-transcription recombinase polymerase amplification. J Virol Methods 222: 138-144.
- Sukal, A., Kidanemariam, D., Dale, J., James, A. and Harding, R. (2017). Characterization of Badnaviruses infecting Dioscorea spp. in the Pacific reveals two putative novel species and the first report of dioscorea bacilliform RT virus 2. Virus Res 238: 29-34.
- Turaki, A. A., Bomer, M., Silva, G., Kumar, P. L. and Seal, S. E. (2017). PCR-DGGE analysis: Unravelling complex mixtures of Badnavirus sequences present in yam germplasm. Viruses 9(7).
- Umber, M., Filloux, D., Muller, E., Laboureau, N., Galzi, S., Roumagnac, P., Iskra-Caruana, M. L., Pavis, C., Teycheney, P. Y. and Seal, S. E. (2014). The genome of African yam (Dioscorea cayenensis-rotundata complex) hosts endogenous sequences from four distinct Badnavirus species. Mol Plant Pathol 15(8): 790-801.
- Umber, M., Gomez, R. M., Gelabale, S., Bonheur, L., Pavis, C. and Teycheney, P. Y. (2017). The genome sequence of Dioscorea bacilliform TR virus, a member of the genus Badnavirus infecting Dioscorea spp., sheds light on the possible function of endogenous Dioscorea bacilliform viruses. Arch Virol 162(2): 517-521.
- Vincze, T., Posfai, J. and Roberts, R. J. (2003). NEBcutter: A program to cleave DNA with restriction enzymes. Nucleic Acids Res 31(13): 3688-3691.
- Yang, I. C., Hafner, G. J., Revill, P. A., Dale, J. L. and Harding, R. M. (2003). Sequence diversity of South Pacific isolates of Taro bacilliform virus and the development of a PCR-based diagnostic test. Arch Virol 148(10): 1957-1968.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Bömer, M., Turaki, A. A., Rathnayake, A. I., Silva, G., Lava Kumar, P. and Seal, S. E. (2018). Rolling Circle Amplification to Screen Yam Germplasm for Badnavirus Infections and to Amplify and Characterise Novel Badnavirus Genomes. Bio-protocol 8(1): e2672. DOI: 10.21769/BioProtoc.2672.
Category
Plant Science > Plant molecular biology > DNA > DNA extraction
Molecular Biology > DNA > PCR
Molecular Biology > DNA > DNA extraction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.