- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

The RiboPuromycylation Method (RPM): an Immunofluorescence Technique to Map Translation Sites at the Sub-cellular Level
Published: Vol 8, Iss 1, Jan 5, 2018 DOI: 10.21769/BioProtoc.2669 Views: 13783
Reviewed by: Alessandro DidonnaJosé M. DiasSalome Calado Botelho
Original research article
The authors used this protocol in:

Apr 2012
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
While isotopic labeling of amino acids remains the reference method in the field for quantifying translation rate, it does not provide any information on spatial localization of translation sites. The rationale behind developing the ribopuromycylation method (RPM) was primarily to map translation sites at the sub-cellular level while avoiding detection of newly synthesized proteins released from ribosomes. RPM visualizes actively translating ribosomes in cells via standard immunofluorescence microscopy in fixed and permeabilized cells using a puromycin-specific monoclonal antibody to detect puromycylated nascent chains trapped on ribosomes treated with a chain elongation inhibitor.
Keywords: Translation siteBackground
For decades, isotopic labeling of amino acids has been considered as the gold standard for studying protein translation. Though this method has proven to be remarkably accurate for evaluating translation rates, it provides no information on the location of translating ribosomes. More recently, amino acids analogs have enabled fluorescent detection nascent chains (Dieterich et al., 2007). Nevertheless, nearly all of the detected signal comes from polypeptides released from ribosome. Our initial idea was to develop a method to label nascent chains while still tethered to translating ribosomes.
Puromycin (PMY) is an aminoglycoside antibiotic that mimics charged tRNATyr and incorporates into the ribosome A site. Consequently, PMY triggers premature translation termination by ribosome catalyzed-covalent incorporation into the nascent chain COOH-terminus (Pestka, 1971) followed by release of PMY-peptide. Polyclonal antibodies (Abs) to PMY were initially generated (Eggers et al., 1997) to detect puromycylated nascent chains released from ribosomes by immunoblotting and immunoprecipitation. Subsequently, fluorescent PMY was used to label nascent chains by microscopy (Starck et al., 2004). Schmidt et al. (2009) found that cells exposed to PMY generate a sufficient amount of PMY-terminated cell surface proteins to enable detection by live cell flow cytometry using a monoclonal Ab (mAb), providing a measure of translation rates. Importantly, none of these methods discriminate between ribosome-attached or released PMY-peptides and are all hindered to some extent as measures of translation by degradation of released proteins.
Having found that chain elongation inhibitors, such as cycloheximide (CHX) or emetine, prevent the release of PMY-nascent chain from ribosomes, we developed the ribopuromycylation method (RPM) that enables immunofluorescent detection of translation sites at the sub-cellular level (Figure 1) (David et al., 2011 and 2012b). This method has been used by our labs and many other labs to study translation in neurons (Biever et al., 2015; Perry et al., 2016; Williams et al., 2016), migrating cells (Willett et al., 2011), immune cells (Seedhom et al., 2016) and stressed or infected cells (Kedersha et al., 2016; Emmott et al., 2017; Roth et al., 2017).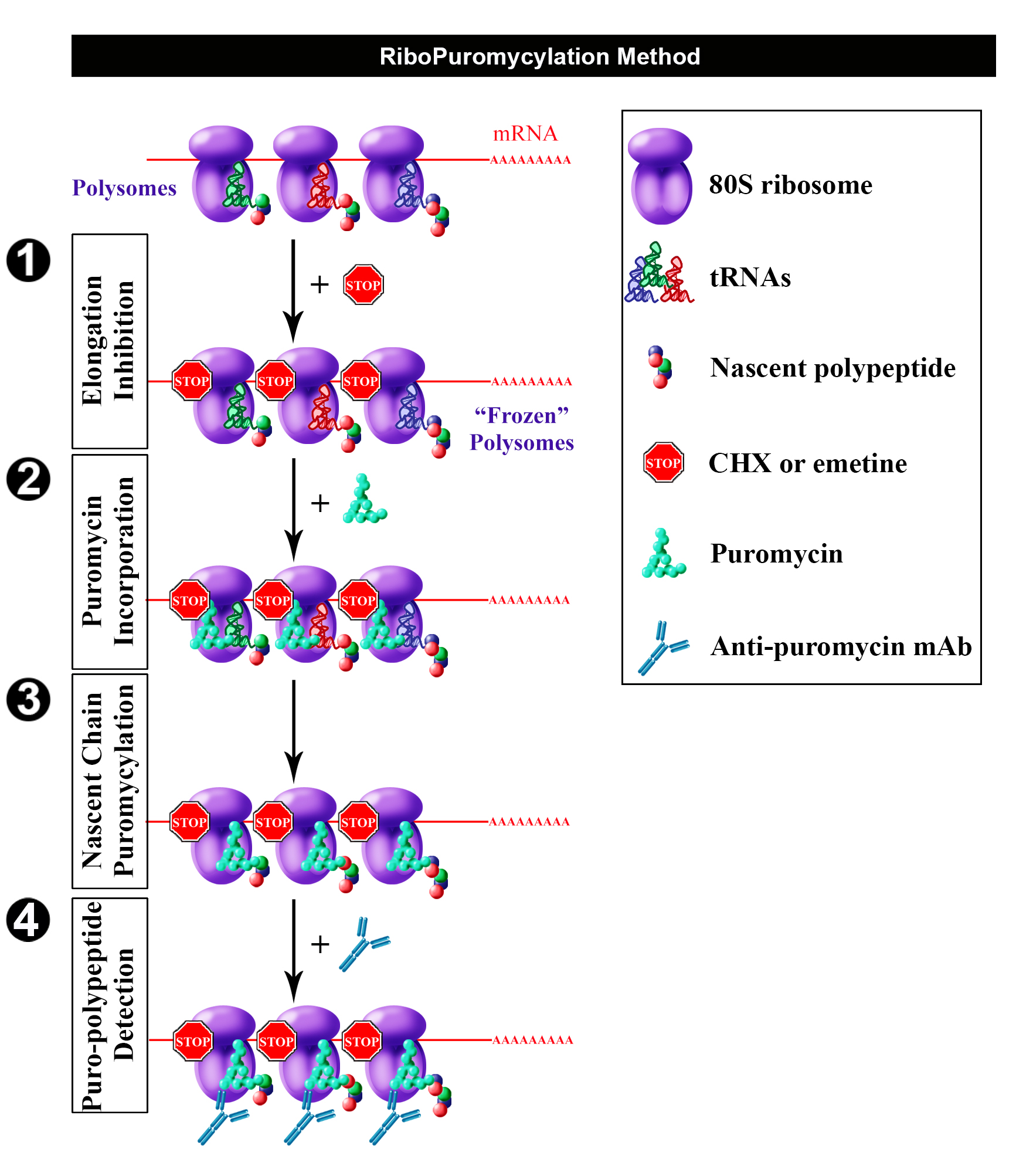
Figure 1. Schematic representation of RPM (from David et al., 2012b). Following freezing of polysome with an elongation inhibitor (step 1), PMY is added (step 2) to living cells and nascent chains become puromycylated through ribosome catalysis (step 3). Anti-PMY monoclonal antibodies detect puromycylated nascent chains via indirect immunofluorescence (step 4). Reproduced from David et al. (2012b) with permission of the publisher and of Dr. Yewdell.
Materials and Reagents
- Materials
- 6-well and 24-well plates (Corning, Costar®, catalog numbers: 3506 , 3524 )
- High quality glass coverslips, 12 mm diameter, #1 thickness (Glaswarenfabrik Karl Hecht, Assistent, catalog number: 1001/12 )
Note: Either autoclave or sterilize with 70% ethanol before use. - Microscope slides (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: J1800AMNZ )
- Whatman paper (GE Healthcare, catalog number: 3030-917 )
- Parafilm (Bemis, catalog number: 701606 )
- Petri dishes (Corning, catalog number: 430591 )
- Aluminum foil (Sigma-Aldrich, catalog number: Z691569 )
Manufacturer: Heathrow Scientific, catalog number: HD23534A . - Plastic tips
- 6-well and 24-well plates (Corning, Costar®, catalog numbers: 3506 , 3524 )
- Cell line(s)
The described procedure and associated figure (Figure 2) uses HeLa cells (ATCC, catalog number: CCL-2.1 )
Note: Though this procedure was originally designed for HeLa cells, it can be easily adapted for other adherent cell lines (Graber et al., 2013). However, controls with several inhibitors are needed to validate any adjustment (e.g., Digitonin concentration) of any kind. For non-adherent cells, we describe an alternate protocol (Procedure D). - Reagents
- Alcian blue (Sigma-Aldrich, catalog number: A5268 )
- 70% ethanol (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R40135 )
- Hoechst 33258 (Thermo Fisher Scientific, InvitrogenTM, catalog number: H1398 )
- Fluoromount-G (SouthernBiotech, catalog number: 0100-01 )
- Potassium phosphate monobasic (KH2PO4)
- Sodium chloride (NaCl)
- Na2HPO4·7H2O
- Dulbecco’s modified Eagle medium (DMEM) (Thermo Fisher Scientific, GibcoTM, catalog number: 41966029 )
- Glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 25030081 )
- Penicillin/streptomycin (Thermo Fisher Scientific, GibcoTM, catalog number: 10378016 )
- Fetal bovine serum (FBS) (Eurobio, catalog number: CVFSVF0101 )
- Digitonin (Wako Pure Chemical Industries, catalog number: 043-21376 )
- Tris-HCl pH 7.5 (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15567027 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M0250 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 )
- Complete Mini EDTA-free protease inhibitor tablets (Sigma-Aldrich, Roche Diagnostics, catalog number: 11836170001 )
- RNase Out (Life Technologies, catalog number: 100000840 ; or Thermo Fisher Scientific, InvitrogenTM, catalog number: 10777019 )
- DEPC treated water (Thermo Fisher Scientific, InvitrogenTM, catalog number: 750023 )
- 16% paraformaldehyde (PFA) (Electron Microscopy Sciences, catalog number: 15710 )
- Sucrose (Sigma-Aldrich, catalog number: 84097 )
- Saponin (Sigma-Aldrich, catalog number: 84510 )
- Glycine (Sigma-Aldrich, catalog number: G7126 )
- Alcian blue (Sigma-Aldrich, catalog number: A5268 )
- Protein synthesis inhibitors (see Table 1)
- Anisomycin (Sigma-Aldrich, catalog number: A9789 )
- Cycloheximide (CHX) (Sigma-Aldrich, catalog number: C7698 )
- Emetine dihydrochloride (Sigma-Aldrich, catalog number: E2375 )
- Harringtonine (Santa Cruz Biotechnology, catalog number: sc-204771 )
- Puromycin (PMY) (Sigma-Aldrich, catalog number: P7255 )
- Sodium arsenite (NaAsO2) (Sigma-Aldrich, catalog number: S7400 )
- Anisomycin (Sigma-Aldrich, catalog number: A9789 )
- Antibodies
- Primary antibodies
- Anti-PMY mouse monoclonal antibodies: We tested 3 mouse mAbs from different hybridoma clones 12D10, 2A4, 5B12. Clone 12D10 was generated by the Pierre laboratory (Schmidt et al., 2009), and is commercially available (Merck, catalog number: MABE343 ). We generated 2A4 and 5B12. 2A4 cells and supernatant are freely available to the scientific community from the Developmental Studies Hybridoma Bank http://dshb.biology.uiowa.edu/PMY-2A4
- Anti-ribosomal P antibody: human polyclonal autoimmune antiserum (from lupus patients) which recognizes three proteins of the 60S ribosomal subunit, RPLP0, P1 and P2 (Immunovision, catalog number: HPO-0100 )
- Anti-lysyl-tRNA synthetase (KRS) antibody: rabbit polyclonal serum which recognizes KRS enzyme (Abcam, catalog number: ab31532 )
- Secondary antibodies
- Donkey anti-mouse Alexa Fluor 488 (Jackson ImmunoResearch, catalog number: 715-545-150 )
- Donkey anti-rabbit Alexa Fluor 594 (Jackson ImmunoResearch, catalog number: 711-585-152 )
- Donkey anti-human Cy5 (Jackson ImmunoResearch, catalog number: 709-175-149 )
- Solutions (see Recipes)
- Anisomycin stock solution (1,000x)
- Cycloheximide (CHX) stock solution (1,000x)
- Emetine dihydrochloride stock solution (1,000x)
- Harringtonine stock solution (1,000x)
- PMY stock solution (1,000x)
- Sodium arsenite stock solution (1,000x)
- Phosphate buffered saline (PBS)
- Growth medium
- Labeling medium
- Labeling control medium
- Extraction buffer
- Wash buffer
- 3% PFA
- Co-extraction/fixation buffer
- Staining buffer (SB)
- Anisomycin stock solution (1,000x)
Equipment
- Ice bucket
- 37 °C water bath (JULABO, model: TW20 )
- Class II laminar flow hood (FASTER, model: SafeFAST Elite )
- Microscopy ultrafine tweezers (Electron Microscopy Sciences, catalog number: 78522-7 )
- PIPETMAN 1-20 µl (Sartorius, model: Proline® Plus, catalog number: 728030 )
- PIPETMAN 20-200 µl (Sartorius, model: Proline® Plus, catalog number: 728060 )
- PIPETMAN 100-1,000 µl (Sartorius, model: Proline® Plus, catalog number: 728070 )
- Slide tray (Glaswarenfabrik Karl Hecht, Assistent, catalog number: 2700/10 )
- Cell Incubator (Heraeus, model: HERACell )
- Centrifuge (Eppendorf, model: 5415 R )
- Microwave
- Microscopy
Laser scanning confocal microscope: Leica TCS SP5 (Leica Microsystems, model: Leica TCS SP5 ) with an HCX PL APO lambda blue 63.0x 1.40 oil UV objective. We used type FF immersion liquid (Cargille-Sacher Laboratories, catalog number: 16212 )
Note: Other comparable systems may be used.
Software
- LAS AF V2.3.1 software (Leica)
- Imaris (Bitplane) and Huygens Essential Software for image deconvolution using the classical maximum likelihood estimation algorithm (V3.6, Scientific Volume Imaging BV, Hilversum, The Netherlands)
- ImageJ (NIH) https://imagej.net/Downloads
Procedure
In the following protocol, we have included controls needed to ensure the specificity of both labeling and staining steps: 3 ‘translation inhibitor controls’ and 1 ‘no PMY’ control. Furthermore, we describe a quadruple staining protocol that permits simultaneous visualization of nuclear translation sites (PMY), large subunit ribosomal proteins (RPLP0, RPLP1, RPLP2), a component of the multi-synthetase complex (lysyl-tRNA synthetase, KRS) and DNA (Hoechst 33258).
As emphasized in ‘Materials and Regents #3 Solutions’, either emetine or CHX can be used as elongation inhibitors to ‘trap’ PMY in translating ribosomes. However, emetine has proven to be more efficient for PMY labeling than CHX (David et al., 2012a). Furthermore, being irreversible, emetine, unlike CHX does not have to be maintained in all solutions throughout the procedure.
Two RPM methods have been developed: the original procedure (Procedure A) and a co-fixation/extraction procedure (Procedure B), easier and more adapted for non-adherent and primary cell cultures. Furthermore, labeling non-adherent cells necessitates an additional step (Procedure C).
- Procedure A (Original RPM protocol)
Day 1- Pre-warm growth medium (see Recipes) in 37 °C water bath. In a cell culture hood, distribute 4 ml of warm growth medium per well in 5 wells of a 6-well plate. In each well, carefully place up to five non-overlapping coverslips using sterile forceps.
Notes:- Backup plan. Coverslips are easily broken and ‘accidents’ frequently happen. Moreover, it’s always interesting to perform RPM with multiple Abs. For these reasons, we usually work with 4 coverslips per well.
- High quality forceps with extremely sharp tips are recommended to enable easy removal of the coverslips from wells. Practice is required to develop the knack of picking them up from 24-well plates without breaking them.
- Avoid coverslip overlapping. In order to be certain that coverslips will not move around in the well and overlap each other: (1) always add the medium first; (2) gently press down the coverslips down to remove any air bubbles between coverslip and the bottom of the well, gently slide the coverslips in order to create an adhesive force.
- Backup plan. Coverslips are easily broken and ‘accidents’ frequently happen. Moreover, it’s always interesting to perform RPM with multiple Abs. For these reasons, we usually work with 4 coverslips per well.
- Transfer 1 ml of growth medium containing 0.5 x 106 HeLa cells per ml in each well. Softly shake the plates to distribute cells uniformly in the wells. Incubate for 24 h to allow HeLa cells to attach tightly to the coverslips and spread. Immunofluorescent resolution is maximized when visualizing well spread cells.
Day 2- Examine cells using an inverted microscope to ensure they have reached 70%-90% confluence. Several solutions must be prepared, at distinct temperatures.
Warm to 37 °C:- 10 ml of growth medium.
- 4 ml of freshly prepared labeling medium (prepared extemporaneously, see Recipes).
- 1 ml of labeling control medium (prepared extemporaneously, see Recipes).
- 40 ml of PBS buffer (see Recipes).
- 5 ml of extraction buffer (freshly made, see Recipes).
- 5 ml of wash buffer (freshly made, see Recipes).
- 10 ml of growth medium.
- Optional step: Ensuring the specificity of PMY labeling necessitates pre-incubation with several translation inhibitors (see Table 1). This step is mandatory when working with new cell lines. The described protocol is designed to accommodate 3 ‘translation inhibitor controls’, preventing PMY labeling in different ways. Dilute each inhibitor in 2 ml of pre-warmed growth media. Assign a well for each: aspirate the media and replace it with media containing the corresponding inhibitor. Incubation times vary depending on the nature of the antibiotic and are indicated in Table 1.
Note: Necessary controls. When performing RPM procedure for the first time (or with a new cell line), we recommend using at least 3 control conditions:- Without ‘active ribosome’, using a translation initiation inhibitor (such as Harringtonine) that results in ribosome run off of mRNA, maximally releasing nascent chains.
- Without ‘ribosome catalyzed PMY incorporation’, i.e., no PMY.
- Using a PMY competitor, such as anisomycin, and in the ‘absence of antigen’, i.e., without PMY labeling.
Table 1. Protein synthesis inhibitors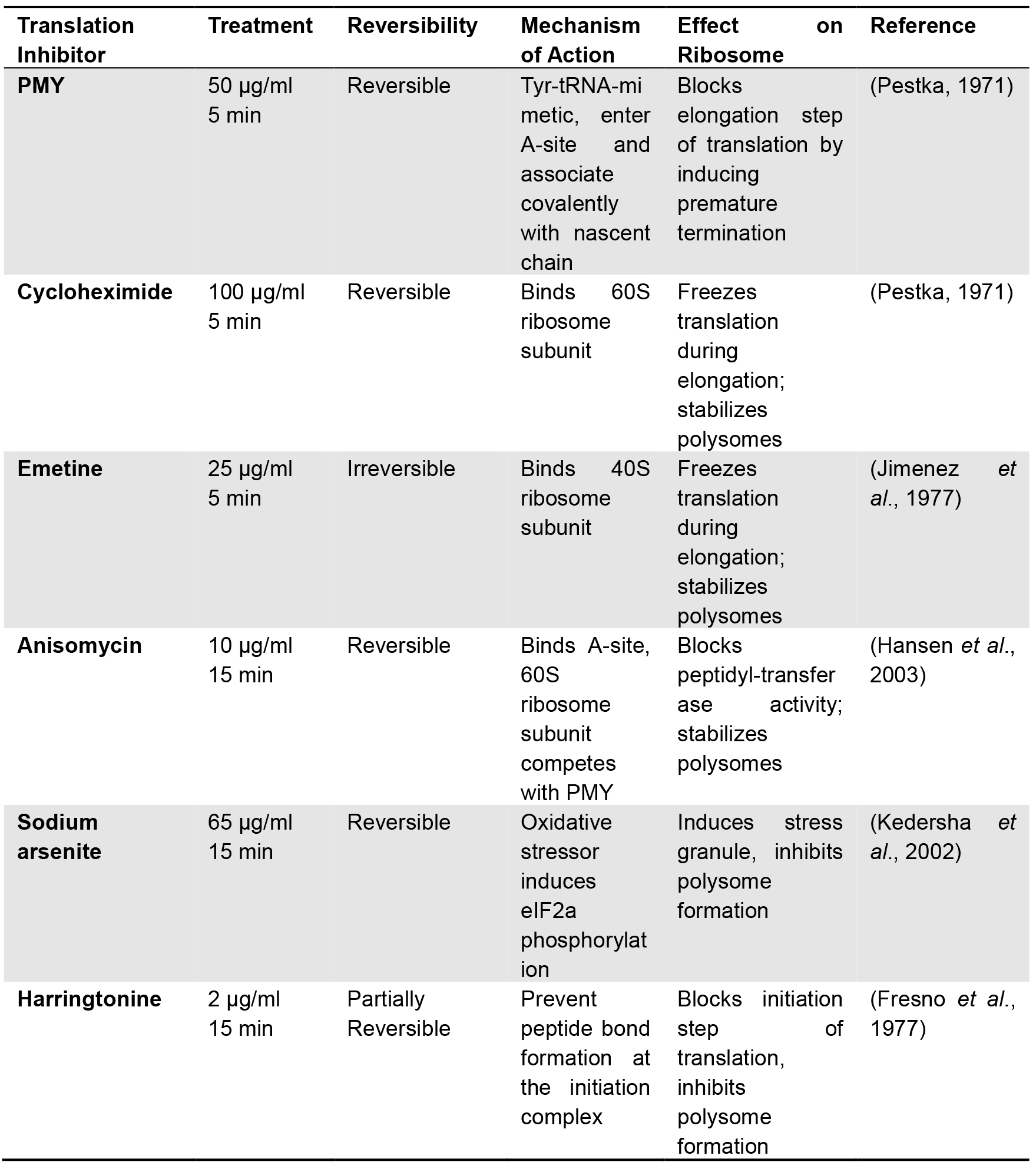
- Without ‘active ribosome’, using a translation initiation inhibitor (such as Harringtonine) that results in ribosome run off of mRNA, maximally releasing nascent chains.
- Aspirate media in each well and replace with 900 µl of pre-warmed labeling medium (test well + 3 ‘inhibitor controls’ wells) or labeling control (in the last well). Incubate for 5 min at 37 °C.
Note: Co-incubation with PMY and elongation inhibitor. Because emetine freezes translation instantly, it can be added simultaneously with PMY.
Reversibility. Anisomycin, being a reversible competitor of PMY, we recommend adding anisomycin in the corresponding well during the PMY labeling incubation (to maintain a constant anisomycin concentration). - Place the 6-well plate on ice, aspirate the medium and wash with 5 ml of ice-cold PBS.
- Aspirate PBS and add 1 ml of ice-cold extraction buffer in each well. Incubate for 2 min on ice.
Note: Be careful with pipetting. It’s crucial from this step to slowly add buffer down the side of the well and avoid detaching the cells. - Aspirate extraction buffer and extremely gently add 900 µl of ice-cold wash buffer.
- Aspirate gently on the side and extremely gently add 900 µl of freshly made 3% PFA. Incubate for 15 min at RT. Then, replace the fixing solution with 2 ml of ice-cold PBS. Check the wells using an inverted microscope to make sure that cells are still attached.
Note: Storage prior to staining. Following PFA fixation, cells may be kept for at least 7 days at 4 °C (in PBS) without noticeably affecting the quality of the RPM staining. Likewise, stained coverslips can be stored for long periods (years, even decades) at -20 °C and retrieved. - Proceed to ‘immunostaining procedure’ (Procedure D).
- Pre-warm growth medium (see Recipes) in 37 °C water bath. In a cell culture hood, distribute 4 ml of warm growth medium per well in 5 wells of a 6-well plate. In each well, carefully place up to five non-overlapping coverslips using sterile forceps.
- Procedure B (co-extraction/fixation procedure)
- Follow Steps 1-6 of the ‘original RPM procedure’ (Procedure A).
- Aspirate PBS and add 1 ml of ice-cold Co-extraction/fixation buffer (see Recipes). Incubate for 20 min on ice.
- Aspirate Co-extraction/fixation buffer.
- Add 900 µl of freshly made 3% PFA. Incubate for 10 min at RT. Then, replace the fixing solution with 2 ml PBS.
- Proceed to ‘immunostaining procedure’ (Procedure D).
- Follow Steps 1-6 of the ‘original RPM procedure’ (Procedure A).
- PMY labeling procedure on non-adherent cells
This procedure should be employed for non-adherent cells such as human peripheral blood monocytes (David et al., 2012b). It permits efficient attachment of cells to coverslips within minutes.- ‘Alcian blue treated coverslips’ must be prepared in advance as followed:
- Prepare a solution of 1% (w/v) Alcian blue (Sigma-Aldrich) in distilled water. This solution lasts for months at RT.
- Completely cover 100-200 coverslips in few ml of this solution in a microwave-safe container.
- Stir manually, making sure that all coverslips are coated with Alcian blue (coverslips tend to stick together).
- Heat for 30 sec to 1 min in the microwave (maximum power intensity, the solution should boil for few seconds).
- Mix again manually, by shaking the container.
- Heat again until boiling.
- Discard Alcian blue solution.
- Wash with distilled water and with a gloved hand dissociate aggregated coverslips.
- Wash with 70% ethanol until only a very light blue shade remains on coverslips.
- Thoroughly wash with water.
- Separate coverslips by hand on a large piece of Whatman paper and let them dry.
- Alcian blue coated coverslips can be kept for months at RT.
- Prepare a solution of 1% (w/v) Alcian blue (Sigma-Aldrich) in distilled water. This solution lasts for months at RT.
- Place one Alcian blue treated coverslip per well of a 24-well plate.
- Centrifuge cells at 400 x g for 5 min at room temperature, wash cells by resuspending them with 5 ml warm DMEM twice in order to remove any trace of FBS.
Note: Do not use FBS with Alcian blue coverslips. Alcian blue binds to negatively charged macromolecules such as glycosaminoglycans. The presence of FBS in the medium would inhibit Alcian blue association with membrane glycoproteins and prevent cell adhesion on coverslip. - Resuspend cells in a small volume of DMEM (at least 106 cells/ml). Spot one drop of cells (about 50 µl) per coverslips.
- Incubate at 37 °C for 5 to 15 min (the more you wait, the better they stretch out).
- Aspirate the medium and replace it with 1 ml of ice-cold PBS.
- Proceed to the ‘Co-extraction/fixation procedure’ (Procedure B), starting with Step B7.
- ‘Alcian blue treated coverslips’ must be prepared in advance as followed:
- Immunostaining procedure
- Transfer one coverslip of each condition into a 24-well plate (save the others at 4 °C). Then incubate cells with 500 µl of staining buffer (SB) (see Recipes) for 15 min at RT. Meanwhile, dilute primary antibodies in staining buffer: anti-PMY mAb (depending on the clone, final concentration varies between 1-4 µg/ml), anti-KRS Abs (1/200) and anti-ribosomal P Abs (1/5,000). To minimize non-specific binding of secondary antibodies, we usually supplement primary antibodies with 5% serum from the species used to generate the secondary antibodies (typically donkey antibodies from Jackson ImmunoResearch).
Note: Staining buffer components. Glycine will quench the fixative properties of PFA. Saponin will facilitate the accessibility of the antibody to some epitopes. FBS can decrease non-specific binding of primary antibodies. - Lay down a small piece of Parafilm. If needed, tape it on the bench. Spot 30 µl of diluted primary antibodies on Parafilm. This step is used to minimize the amount of primary Abs needed for staining. In 24-well plates, 200 µl is needed to completely cover the coverslip. Staining in 24-well plates will reduce the effort required and minimize errors. While antibodies can be reused, mAbs are generally available in essentially unlimited amounts if you have the hybridoma.
- Using forceps, carefully remove the coverslip, remove excess staining buffer by gently blotting coverslip edge on a Kimwipe and place cells side down on the primary antibody drop on Parafilm. Cover with a Petri dish with a moist paper towel attached to the inner top and incubate at room temperature for 60 min.
Notes:- Staining with ‘precious’ antibody. In order to limit the use of precious antibodies you can spot only 10 µl. In this case, you definitely need to incubate in a ‘moist chamber’ to prevent coverslips from drying out.
- When you inadvertently drop the coverslip. Inevitably, you will drop coverslips and will need to determine the ‘cell side’. This can be done if cells are sufficiently dense by holding up the light and looking for the side with a white film. If unsure, you can scratch the suspected side and see the loss of cells, or place the coverslip on an inverted microscope.
- Staining with ‘precious’ antibody. In order to limit the use of precious antibodies you can spot only 10 µl. In this case, you definitely need to incubate in a ‘moist chamber’ to prevent coverslips from drying out.
- Meanwhile, dilute secondary antibodies in staining buffer: 1/500 for goat anti-mouse A488, 1/500 for donkey anti-rabbit Alexa Fluor 594, 1/500 for donkey anti-human Cy5.
- With forceps, remove coverslips from primary antibody spots, and place in 24-wells plate. Wash three times with 1 ml 1x PBS.
- Lay down a small piece of Parafilm. If needed, tape it on the bench. Spot 30 µl of diluted secondary antibodies solution on Parafilm.
- Using forceps, carefully remove the coverslip from the plate, remove excess wash buffer by gently blotting coverslip edge on a Kimwipe and place cells side down on the secondary antibody drop on Parafilm, cover Petri dish with aluminum foil (to protect the fluorophore), incubate for 45 min at room temperature.
- With forceps, pick up coverslips from secondary antibody spots, and place them in a 24-well plate. Wash twice with 1 ml 1x PBS. Then wash again with 1 ml distilled water. Dilute Hoechst 33258 in distilled water (1 µg/ml).
Note: PBS does not solubilize Hoechst 33285. For this reason, we recommend using distilled water for washes after this step. - Aspirate distilled water and add 200 µl of diluted Hoechst solution. Incubate for 5 min at RT.
- Aspirate and wash twice with distilled water.
- Place a drop of Fluoromount-G (5 µl) on slides.
Notes:- Fluoromount-G. This mounting solution is quite viscous when cold. To facilitate the pipetting of small volumes (5 µl), we usually warm up the solution at RT for 15 min before using. Another helpful trick: cutting the tip of the plastic tip with a razor or scissor helps.
- The number of coverslips per slide. With practice, up to 8 coverslips can be placed on each slide. It is greatly advantageous to minimize the number of slides that have to be manipulated during microscopy which should be performed in the dark to accommodate the eyes and maximize visual acuity. Generally, the focal plane only has to be established one time for each slide, allowing rapid viewing of coverslips on the same slide. Give careful thought to the order of the coverslips on the slide. Put the most important coverslips for comparison with each other as closely as possible on the slide. The most important antibodies should be visualized with colors that can be seen by eye. It is important to form a general impression of staining of as many cells as possible, and this is by far most easily done by eye. Equally important is to write down your conclusions of the staining during or immediately after viewing the slides.
- Fluoromount-G. This mounting solution is quite viscous when cold. To facilitate the pipetting of small volumes (5 µl), we usually warm up the solution at RT for 15 min before using. Another helpful trick: cutting the tip of the plastic tip with a razor or scissor helps.
- Using forceps carefully pry up an edge and remove the coverslip, gently place cell side down on a Kimwipe to remove water and then place cells side down on mounting solution drops.
- Place slides in a tray and leave them dry overnight at room temperature in a drawer to protect the fluorophore.
Note: Fast dry. If needed, drying may be hastened by incubating slides at 37 °C for 2-3 h. Never examine slides before mountant is dry, as this can damage the extremely expensive oil immersion objectives. - Store the tray at 4 °C until analysis. Staining is stable for at least 2 weeks at 4 °C in darkness. For a longer storage we recommend using slide boxes and storing at -20 °C.
- Analyze with a confocal microscope (Figure 2).
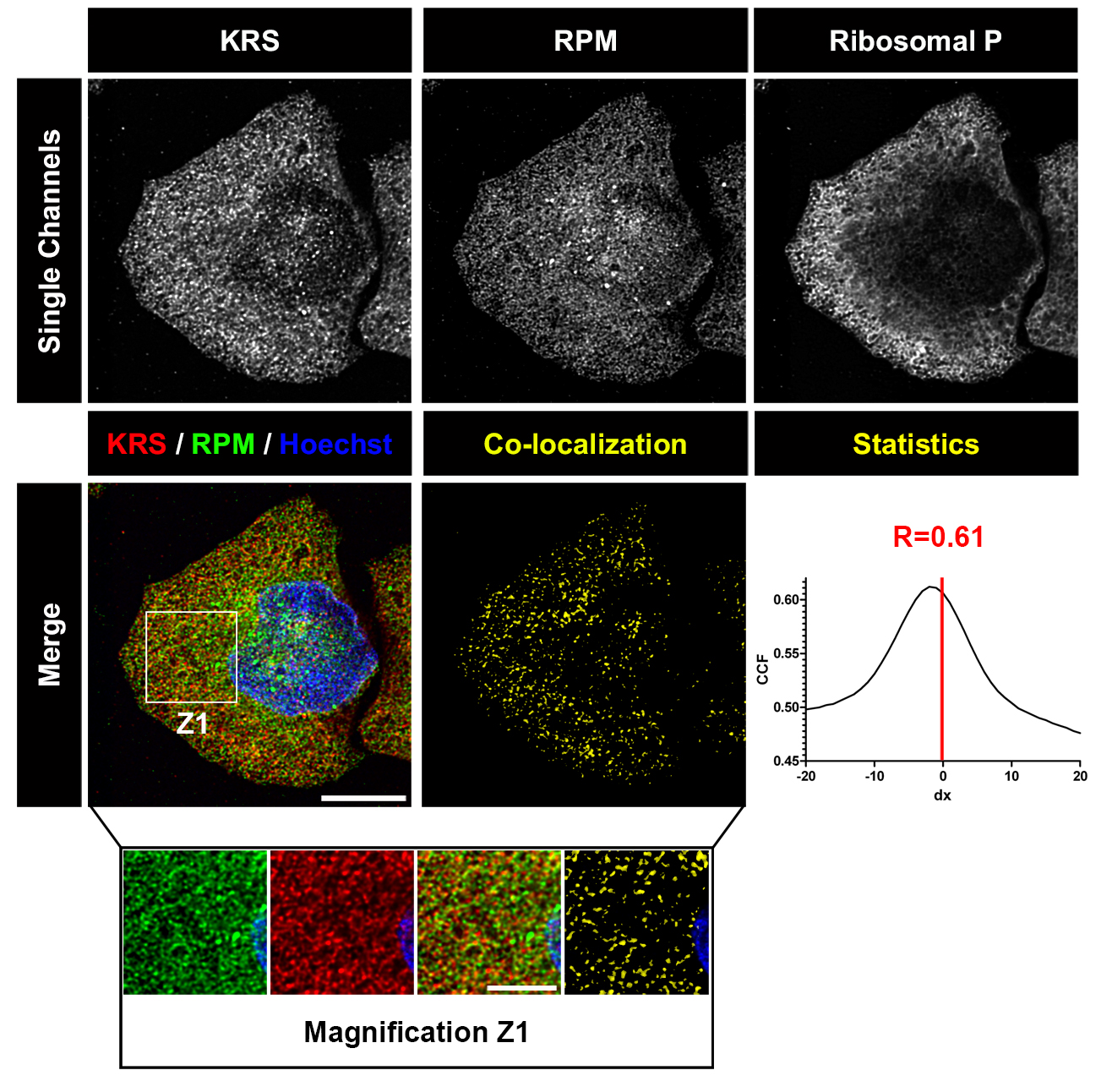
Figure 2. Deconvolved images of HeLa cell labeled with RPM (from David et al., 2011). HeLa cells were pulsed with PMY + CHX to label translating ribosomes and extracted with Dig to remove free PMY and cytosolic components. Cells were then fixed, permeabilized and stained for KRS, PMY or ribosomal P proteins. Co-localization was estimated using ImageJ (NIH) and JACoP plugin that compiles general co-localization indicators such as Pearson’s coefficient (Manders et al., 1992) and Van Steensel’s CCF (Van Steensel et al., 1996). KRS and RPM demonstrate extensive co-localization as quantitated by Van Steensel’s CCF greater than 0.75 and Pearson’s coefficient (R) greater than 0.5. Bar scales, 10 μm, 5 μm for Z1. Reproduced from David et al., 2011 with permission of the publisher and of Dr. Yewdell.
- Transfer one coverslip of each condition into a 24-well plate (save the others at 4 °C). Then incubate cells with 500 µl of staining buffer (SB) (see Recipes) for 15 min at RT. Meanwhile, dilute primary antibodies in staining buffer: anti-PMY mAb (depending on the clone, final concentration varies between 1-4 µg/ml), anti-KRS Abs (1/200) and anti-ribosomal P Abs (1/5,000). To minimize non-specific binding of secondary antibodies, we usually supplement primary antibodies with 5% serum from the species used to generate the secondary antibodies (typically donkey antibodies from Jackson ImmunoResearch).
Data analysis
Images can be processed using LAS AF software (Leica), Imaris (Bitplane), Huygens Essentials Software (Version 3.6, Scientific Volume Imaging BV), Photoshop CS2 (Adobe), and/or ImageJ. For obvious ethical reasons, gamma function–which connects the numerical value of a pixel with its actual luminance–must not be manipulated. Each set of images for a given experiment must be processed identically to maintain the image intensity ratio. In previous publications (David et al., 2011; 2012a and 2012b; Macari et al., 2015), ImageJ and Prism software were used for quantitation and statistical analysis. Comparing translation activity from different condition necessitates acquisition of multiple fields (at least 6 fields per condition for statistical significance). In order to normalize each field, the mean fluorescence ratio of PMY/ribo P staining must be quantitated using ImageJ. Then, values may be plotted (mean ± SEM). For statistical analysis, we previously used two-tailed unpaired t-test. An example is presented in the following paper: David et al., 2012.
Notes
As stated above (methods #4), multiple controls are necessary when applying RPM for the first time or using new cell lines.
Recipes
- Anisomycin (Calbiochem) stock solution (1,000x)
10 mg/ml (or 37 mM) in 100% ethanol solution
Store at -20 °C - Cycloheximide (CHX) stock solution (1,000x)
100 mg/ml (or 355 mM) in 50% ethanol
Store at -20 °C - Emetine dihydrochloride stock solution (1,000x)
25 mg/ml (or 45 mM) in 50% ethanol
Store at -20 °C - Harringtonine stock solution (1,000x)
2 mg/ml (or 3.7 mM) in 100% ethanol
Store at -20 °C - PMY stock solution (1,000x)
50 mg/ml (or 91 mM) in 50% ethanol
Store at -20 °C - Sodium arsenite stock solution (1000x)
65 mg/ml (or 500 mM) in distilled water
Store at 4 °C - Phosphate buffered saline (PBS)
210.0 mg/L KH2PO4
9,000 mg/L NaCl
726.0 mg/L Na2HPO4·7H2O - Growth medium
Dulbecco’s modified Eagle’s medium with:
Glutamine 2 mM final
1% penicillin/streptomycin
7.5% fetal bovine serum (FBS) - Labeling medium
Add 5 µl of PMY stock solution (91 µM final) and either 5 µl of emetine stock solution (45 µM final) or 5 µl of CHX stock solution (355 µM final) to 5 ml growth medium - Labeling control medium
Add either 5 µl of emetine stock solution (45 µM final) or 5 µl of CHX stock solution (355 µM final) to 1 ml growth medium - Extraction buffer
0.015% (m/v) digitonin
50 mM Tris-HCl pH 7.5
5 mM MgCl2
25 mM KCl
355 µM CHX
1x EDTA-free protease inhibitors (1 tablet per 10 ml)
10 U/ml RNase Out
DEPC treated water - Wash buffer
50 mM Tris-HCl pH 7.5
5 mM MgCl2
25 mM KCl
355 µM CHX
1x EDTA-free protease inhibitors (1 tablet per 10 ml)
10 U/ml RNase Out
DEPC treated water - 3% PFA
Dilute stock solution (16%) in 1x PBS - Co-extraction/fixation buffer
0.015% (m/v) digitonin
50 mM Tris-HCl pH 7.5
5 mM MgCl2
25 mM KCl
0.2 M sucrose
355 µM CHX
1x EDTA-free protease inhibitors (1 tablet per/10 ml)
10 U/ml RNase Out
3% PFA
DEPC treated water - Staining buffer (SB)
0.05% saponin
10 mM glycine
5% FBS
1x PBS
Acknowledgments
JWY is generously supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases. AD benefits from generous funding from Fondation pour la Recherche Médicale, Ligue contre le Cancer and Cancéropôle GSO. The authors declare no conflicts of interest or competing interests.
References
- Biever, A., Puighermanal, E., Nishi, A., David, A., Panciatici, C., Longueville, S., Xirodimas, D., Gangarossa, G., Meyuhas, O., Herve, D., Girault, J. A. and Valjent, E. (2015). PKA-dependent phosphorylation of ribosomal protein S6 does not correlate with translation efficiency in striatonigral and striatopallidal medium-sized spiny neurons. J Neurosci 35(10): 4113-4130.
- David, A., Bennink, J. R. and Yewdell, J. W. (2012a). Emetine optimally facilitates nascent chain puromycylation and potentiates the ribopuromycylation method (RPM) applied to inert cells. Histochem Cell Biol 139(3): 501-504.
- David, A., Dolan, B. P., Hickman, H. D., Knowlton, J. J., Clavarino, G., Pierre, P., Bennink, J. R. and Yewdell, J. W. (2012b). Nuclear translation visualized by ribosome-bound nascent chain puromycylation. JCB 197(1): 45-57.
- David, A., Netzer, N., Strader, M. B., Das, S. R., Chen, C. Y., Gibbs, J., Pierre, P., Bennink, J. R. and Yewdell, J. W. (2011). RNA binding targets aminoacyl-tRNA synthetases to translating ribosomes. J Biol Chem 286(23): 20688-20700.
- Dieterich, D. C., Lee, J. J., Link, A. J., Graumann, J., Tirrell, D. A. and Schuman, E. M. (2007). Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat Protoc 2(3): 532-540.
- Eggers, D. K., Welch, W. J. and Hansen, W. J. (1997). Complexes between nascent polypeptides and their molecular chaperones in the cytosol of mammalian cells. Mol Biol Cell 8(8): 1559-1573.
- Emmott, E., Sorgeloos, F., Caddy, S. L., Vashist, S., Sosnovtsev, S., Lloyd, R., Heesom, K., Locker, N. and Goodfellow, I. (2017). Norovirus-mediated modification of the translational landscape via virus and host-induced cleavage of translation initiation factors. Mol Cell Proteomics 16(4 suppl 1): S215-S229.
- Fresno, M., Jimenez, A. and Vazquez, D. (1977). Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem 72(2): 323-330.
- Graber, T. E., Hebert-Seropian, S., Khoutorsky, A., David, A., Yewdell, J. W., Lacaille, J. C. and Sossin, W. S. (2013). Reactivation of stalled polyribosomes in synaptic plasticity. Proc Natl Acad Sci U S A 110(40): 16205-16210.
- Hansen, J. L., Moore, P. B. and Steitz, T. A. (2003). Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol 330(5): 1061-1075.
- Jimenez, A., Carrasco, L. and Vazquez, D. (1977). Enzymic and nonenzymic translocation by yeast polysomes. Site of action of a number of inhibitors. Biochemistry 16(21): 4727-4730.
- Kedersha, N., Chen, S., Gilks, N., Li, W., Miller, I. J., Stahl, J. and Anderson, P. (2002). Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13(1): 195-210.
- Kedersha, N., Panas, M. D., Achorn, C. A., Lyons, S., Tisdale, S., Hickman, T., Thomas, M., Lieberman, J., McInerney, G. M., Ivanov, P. and Anderson, P. (2016). G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Biol 212(7): 845-860.
- Macari, F., El-Houfi, Y., Boldina, G., Xu, H., Khoury-Hanna, S., Ollier, J., Yazdani, L., Zheng, G., Bieche, I., Legrand, N., Paulet, D., Durrieu, S., Bystrom, A., Delbecq, S., Lapeyre, B., Bauchet, L., Pannequin, J., Hollande, F., Pan, T., Teichmann, M., Vagner, S., David, A., Choquet, A. and Joubert, D. (2016). TRM6/61 connects PKCα with translational control through tRNAi(Met) stabilization: impact on tumorigenesis. Oncogene 35(14): 1785-1796.
- Manders, E., Stap, J., Brakenhoff, G., van Driel, R. and Aten, J. (1992). Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 103: 857-862.
- Perry, R. B., Rishal, I., Doron-Mandel, E., Kalinski, A. L., Medzihradszky, K. F., Terenzio, M., Alber, S., Koley, S., Lin, A., Rozenbaum, M., Yudin, D., Sahoo, P. K., Gomes, C., Shinder, V., Geraisy, W., Huebner, E. A., Woolf, C. J., Yaron, A., Burlingame, A. L., Twiss, J. L. and Fainzilber, M. (2016). Nucleolin-mediated RNA localization regulates neuron growth and cycling cell size. Cell Rep 16(6): 1664-1676.
- Pestka, S. (1971). Inhibitors of ribosome functions. Annu Rev Microbiol 25: 487-562.
- Roth, H., Magg, V., Uch, F., Mutz, P., Klein, P., Haneke, K., Lohmann, V., Bartenschlager, R., Fackler, O. T., Locker, N., Stoecklin, G. and Ruggieri, A. (2017). Flavivirus infection uncouples translation suppression from cellular stress responses. MBio 8(1).
- Schmidt, E. K., Clavarino, G., Ceppi, M. and Pierre, P. (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6(4): 275-277.
- Seedhom, M. O., Hickman, H. D., Wei, J., David, A. and Yewdell, J. W. (2016). Protein translation activity: A new measure of host immune cell activation. J Immunol 197(4): 1498-1506.
- Starck, S. R., Green, H. M., Alberola-Ila, J. and Roberts, R. W. (2004). A general approach to detect protein expression in vivo using fluorescent puromycin conjugates. Chem Biol 11(7): 999-1008.
- Van Steensel, B., van Binnendijk, E., Hornsby, C., van der Voort, H., Krozowski, Z., de Kloet, E. and van Driel, R. (1996). Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J Cell Sci 109: 787-792.
- Willett, M., Brocard, M., Davide, A. and Morley, S. J. (2011). Translation initiation factors and active sites of protein synthesis co-localize at the leading edge of migrating fibroblasts. Biochem J 438(1): 217-227.
- Williams, K. R., McAninch, D. S., Stefanovic, S., Xing, L., Allen, M., Li, W., Feng, Y., Mihailescu, M. R. and Bassell, G. J. (2016). hnRNP-Q1 represses nascent axon growth in cortical neurons by inhibiting Gap-43 mRNA translation. Mol Biol Cell 27(3): 518-534.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bastide, A., Yewdell, J. W. and David, A. (2018). The RiboPuromycylation Method (RPM): an Immunofluorescence Technique to Map Translation Sites at the Sub-cellular Level. Bio-protocol 8(1): e2669. DOI: 10.21769/BioProtoc.2669.
- David, A., Dolan, B. P., Hickman, H. D., Knowlton, J. J., Clavarino, G., Pierre, P., Bennink, J. R. and Yewdell, J. W. (2012b). Nuclear translation visualized by ribosome-bound nascent chain puromycylation. JCB 197(1): 45-57.
Category
Neuroscience > Cellular mechanisms > Intracellular signalling
Cell Biology > Cell imaging > Fixed-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.