- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Assessing Long-distance Transport from Photosynthetic Source Leaves to Heterotrophic Sink Organs with [14C]CO2
Published: Vol 7, Iss 24, Dec 20, 2017 DOI: 10.21769/BioProtoc.2657 Views: 6605
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Phloem loading and transport of photoassimilate from photoautotrophic source leaves to heterotrophic sink organs are essential physiological processes that help the disparate organs of a plant function as a single, unified organism. We present three protocols we routinely use in combination with each other to assess (1) the relative rates of sucrose (Suc) loading into the phloem vascular system of mature leaves (Yadav et al., 2017a), (2) the relative rates of carbon loading and transport through the phloem (Yadav et al., 2017b), and (3) the relative rates of carbon unloading into heterotrophic sink organs, specifically roots, after long-distance transport (this protocol). We propose that conducting all three protocols on experimental and control plants provides a reliable comparison of whole-plant carbon partitioning, and minimizes ambiguities associated with a single protocol conducted in isolation (Dasgupta et al., 2014; Khadilkar et al., 2016). In this protocol, [14C]CO2 is photoassimilated in source leaves and phloem loading and transport of the 14C label to heterotrophic sink organs, particularly roots, is quantified by scintillation counting. Using this protocol, we demonstrated that overexpression of sucrose transporters and a vacuolar proton pumping pyrophosphatase in the companion cells of Arabidopsis enhanced transport of 14C label photoassimilates to sink organs (Dasgupta et al., 2014; Khadilkar et al., 2016). This method can be adapted to quantify long-distance transport in other plant species.
Keywords: ArabidopsisBackground
Long-distance transport through the phloem from autotrophic source organs to heterotrophic sinks is fundamental to plant growth and yield. Based on its role and its location in the plant and its prevailing function in that location, the phloem network is commonly divided into the collection phloem, the transport phloem, and the release phloem (Ayre, 2011). The collection phloem is where sugars and other compounds are loaded into the phloem in preparation for transport. In established plants, the collection phloem is the minor veins of mature, photoautotrophic leaves where phloem loading occurs. Our first companion protocol (Yadav et al., 2017a) describes how [14C]Suc is used to quantify phloem loading capacity in leaf disks. The transport phloem connects source and sink tissues and represents the longest contiguous stretch of phloem in the long-distance-transport pathway. All photoassimilate destined for the heterotrophic sinks moves along the transport phloem, but the transport phloem is far from a simple pipe connecting regions where most of the loading and unloading occurs. Lateral tissues require nutrients and also act as transient storage reserves in stems and roots such that exchange between the transport phloem and adjacent tissue is highly dynamic (discussed at length in Ayre, 2011). Our second companion protocol (Yadav et al., 2017b) describes how 14C labeling with [14C]CO2 can be coupled with collecting phloem exudates into EDTA solutions to measure photoassimilate loaded into the collection phloem and moving through the transport phloem. The release phloem generally refers to that in terminal sink tissues at or very near the end of the phloem network where rates of unloading are highest. The release phloem is found in regions of rapid cell division and growth, or in storage organs, where resources are most strongly needed. The unloading mechanism from the phloem tissue itself is commonly through plasmodesmata into symplastic domains within the recipient tissue. Subsequent transport across membranes to the apoplast, followed by uptake into adjacent cells, occurs in some organs. Symplastic domains and apoplastic boundaries have been elegantly demonstrated with the green fluorescent protein, which moves readily through apical tips and ovule integuments when unloaded from the release phloem, but does not enter the filial tissue of seeds, which is symplastically isolated from maternal tissues (Stadler et al., 2005a; Stadler et al., 2005b). Here we describe a protocol for photosynthetically labeling source leaves with [14C]CO2 and measuring transport to heterotrophic sink organs, specifically roots. An advantage of this protocol is that it takes a whole-plant, holistic approach to quantifying source to sink relationships between control and experimental plants. A disadvantage is that it does not provide information on the individual steps of the transport process.
Materials and Reagents
- Microcentrifuge tubes, screw cap with O-rings (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3464 )
- Pasteur pipette
- [14C]CO2 labeling chambers derived from square Petri dishes (100 x 100 x 15 mm, Fisher Scientific, catalog number: FB0875711 A; or larger 120 x 120 mm, Fisher Scientific, catalog number: 07-000-330 )
- Sterile scalpel
- Circular deep-well Petri dishes for germinating seeds (100 x 25 mm) (Fisher Scientific, catalog number: FB0875711 )
- Surgical white porous tape (3M, catalog number: 1530-0 )
- 10 ml syringe (without a needle attached) (Luer-Lok Tip) (BD, catalog number: 309604 )
- 1 ml plastic syringe barrel (Luer-Lok Tip) (BD, catalog number: 309628 ), cut to 3 cm, and plunger
- Syringe needle, 1.5-2.0 inches, 18 gauge (Monoject Needle, Covidien, catalog number: 1188818112 )
- Scintillation vials (Fisher Scientific, catalog number: 03-337-15 )
- Double edge razor blades (PERSONNA brand, Electron Microscopy Sciences, catalog number: 72000 )
- Modeling clay (American Art Clay)
- Control and experimental plant material
- Sodium bicarbonate [14C]NaHCO3 (MP Biomedicals, catalog number: 0117441H ; 40-60 mCi/mmol; 2 mCi/ml; 5 mCi; 185 MBq)
- Sodium hypochlorite (NaClO) (Commercial bleach)
- Dow Corning high vacuum grease
- Lactic acid (85%) (Fisher Scientific, catalog number: A162-500 )
- Soda lime (LI-COR, catalog number: 9964-090 ) in a column (e.g., Bio-Rad Econo-Column, 1.5 x 20 cm, Bio-Rad Laboratories, catalog number: 7371522 )
- Ethanol absolute (Pharmco-AAPER, catalog number: 111000200 )
- Murashige and Skoog (MS) medium with Gamborg vitamins (PhytoTechnology Laboratories, catalog number: M404 ) or other synthetic, sterile medium suitable for plant growth in vertically-oriented Petri plates (see Recipes for seed germination and experiments)
- Sucrose (Sigma-Aldrich, catalog number: S0389 )
- Potassium hydroxide (Fisher Scientific, catalog number: P250-500 )
- Gellan gum (PhytoTechnology Laboratories, catalog number: G434 )
- Ecolume scintillation fluid (MP Biomedicals, catalog number: 0188247004 )
- Half-strength Murashige and Skoog (MS) medium with Suc for seed germination (see Recipes)
- Half-strength Murashige and Skoog (MS) medium without Suc for experiments (see Recipes)
Equipment
- Glassware, balance, stir plates, pH meter, autoclave, water bath, clean bench, etc., for sterile media
- Desiccator (2.5 L) for sterilizing seeds (Kimble, catalog number: 31200-150 )
- Environmental growth chambers for growing control and experimental plants
- Personal safety equipment: lab coat, nitrile gloves (or similar), and eye protection
- Lamp suitable for photosynthetic labeling, such as a 400 W metal halide lamp (SYLVANIA 64490 - 400 Watt - BT37 - Metal Halide)
- Fume hood with appropriate support for metal halide lamp (see Figure 1, Yadav et al., 2017b,)
- Slim line micro blowers for air circulation (optional; Exton PA, Pelonis Technologies, catalog number: RFB3004 ; powered by four 1.5 volt D-cell batteries in sequence to provide 6 volts)
- Small vacuum pump with inlet and outlet (e.g., Airpo, Barcodable, catalog number: UPC 045635496699 )
- Geiger counter (Ludlum Measurements, model: Model 3 )
- Scissors (Fisher Scientific, catalog number: 08-951-20 )
- Scalpel handle (Fine Science Tools, catalog number: 10003-12 )
- Scalpel blades (Fine Science Tools, catalog number: 10011-00 )
- Forceps, fine, such as Dumont fine point No. 5 (Fine Science Tools, catalog number: 11251-10 )
- Balance (METTLER TOLEDO, model: AE100 )
- Microcentrifuge (GeneMate, catalog number: C-1301-PC )
- Rotary platform shaker (Orbital Shaker Variable, BioExpress, GeneMate, catalog number: S-3200-LS )
- Scintillation counter (Beckman Counter, model: LS 6000IC )
- Water bath
Procedure
- Preparing a work area suitable for [14C]CO2 photoassimilation
- Refer to Yadav et al., 2017b for instructions on preparing a work area suitable for labeling by [14C]CO2 photoassimilation.
- [14C]NaHCO3 stocks will release gaseous [14C]CO2. To minimize this, commercial stocks are supplied as basic solutions buffered to pH 9.5 since acidic pH promotes conversion to CO2. Stocks should be stored at 4 °C and not -20 °C to prevent freezing and localized concentrations of [14C]NaHCO3 among the water crystals. We recommend aliquoting stock into screw cap microcentrifuge tubes with O-rings. Receipt and use of stocks should be recorded as required by the institute where the experiments are conducted.
- Refer to Yadav et al., 2017b for instructions on preparing a work area suitable for labeling by [14C]CO2 photoassimilation.
- Germination of experimental and control WT Arabidopsis thaliana seeds
- Aliquot 30-40 seeds (~1 mg) of each experimental and control line into separate 2 ml microcentrifuge tubes and sterilize by standard procedures with liquid bleach in the liquid or gas phase. Examples of experimental lines might be transgenic plants with modified transporter gene expression, in which case control lines would be wild type plants or preferably transgenic plants with the same T-DNA backbone but without modified transporter expression. We usually perform gas phase sterilization (Clough and Bent, 1998): in a fume hood, place open microcentrifuge tubes with seeds in a 2.5 L glass desiccator along with a beaker holding 40 ml commercial bleach. Quickly acidify the bleach with concentrated HCl (1-2 ml in a Pasteur pipette with rubber bulb) and seal the desiccator lid in place. Allow seeds to sterilize in the released chlorine gas for 4-5 h. Sprinkle the seeds immediately on germination medium (½ strength MS medium with Gamborg vitamins containing 1% sucrose with 5 g/L gellan gum).
Notes:- It is crucial to avoid uneven distribution of seeds on germination plates, since crowding can influence early seedling growth and development. We sprinkle seeds manually to achieve even distribution.
- Leaving the seeds in chlorine gas for too long can kill the seeds, and seeds sterilized by this method do not store well. Sucrose in the medium improves germination and consistent seedling establishment; later steps use medium without sucrose.
- It is crucial to avoid uneven distribution of seeds on germination plates, since crowding can influence early seedling growth and development. We sprinkle seeds manually to achieve even distribution.
- Stratify the seeds for 3 days at 4 °C in darkness. For germination, transfer plates to the growth chamber under a 12 h light (22 °C)/12 h dark (20 °C) diurnal cycle at 130 μmol photons m-2 sec-1 in the vertical orientation. To avoid contamination, keep the plates sealed during germination.
- Prepare sterile ½ strength MS medium with Gamborg vitamins and 5 g/L gellan gum without sucrose in square culture plates for growth in the vertical orientation. Fill the plates half full with media and once solidified, use a sterile scalpel to aseptically remove 1 cm of medium from one edge of the plate. When placed vertically, the rosettes of each plant will be above this cut so they do not contact the medium, and the roots will lay along the surface of the medium. Medium without sucrose is used to best mimic physiological source and sink relationships.
- Approximately 4-5 days post germination, when roots are ~1 cm long, transfer young seedlings from the germination plates to the square plates. With fine forceps (e.g., Dumont #5), gently lift the seedlings from under the cotyledons without pinching or piercing the hypocotyl and without damaging the root. Lay the root down 3-4 cm below the cut edge of the medium in the square plates and drag the seedling up from under the cotyledons to straighten the roots. Position the seedling such that the root remains on the medium and the hypocotyl and rosette are above the cut edge of the medium.
Note: Each square culture dish will be an independent [14C]CO2 labeling chamber. Therefore, control and experimental plants should be grown together in the same chamber. In 100 x 100 mm plates, we typically grow nine plants: 3 control plants and 3 each of 2 experimental lines with the order alternating in 6 replicate plates. Larger plates will accommodate more plants. Labeling chambers can also be made from containers, such as sterilized deli containers (Yadav et al., 2017b). - Seal the square plates using porous surgical tape and arrange the plates near to vertical (~15° off vertical) to keep the roots growing down, but also in contact to the medium surface. Arrange the plates in a growth chamber with the same conditions described above. Depending on the experiment, labeling with [14C]CO2 will be performed in 4 to 7 days, when the roots have extended ~3 cm to cover ~75-80% the length of the medium, or later, if larger plates are used or if growth is slow. Labeling should be done before the roots reach the bottom of the plate.
- Aliquot 30-40 seeds (~1 mg) of each experimental and control line into separate 2 ml microcentrifuge tubes and sterilize by standard procedures with liquid bleach in the liquid or gas phase. Examples of experimental lines might be transgenic plants with modified transporter gene expression, in which case control lines would be wild type plants or preferably transgenic plants with the same T-DNA backbone but without modified transporter expression. We usually perform gas phase sterilization (Clough and Bent, 1998): in a fume hood, place open microcentrifuge tubes with seeds in a 2.5 L glass desiccator along with a beaker holding 40 ml commercial bleach. Quickly acidify the bleach with concentrated HCl (1-2 ml in a Pasteur pipette with rubber bulb) and seal the desiccator lid in place. Allow seeds to sterilize in the released chlorine gas for 4-5 h. Sprinkle the seeds immediately on germination medium (½ strength MS medium with Gamborg vitamins containing 1% sucrose with 5 g/L gellan gum).
- Photosynthetic labeling
- Photograph the plates to record growth on the day they are to be labeled. Turn on the 400 W metal halide light in the fume hood about an hour before labeling, so it reaches a stable intensity of ~130 μmol photons m-2 sec-1 at the working surface.
- Acclimate the plants under the metal halide lamp for about 30 min before labeling. Remove the surgical tape, but leave the lids in place.
Note: Photosynthesis, carbon partitioning into different metabolic pools, and long-distance transport to sink organs fluctuate through the diurnal cycle. For consistency, we generally label 6 h into the illuminated period. Six plates can be arranged under the light and receive equal light intensity, but care must be taken to ensure that the number of plates labeled does not exceed the ability to process the plant material efficiently in subsequent steps. Table 1 provides a scheduling template.
For each plate to be labeled, prepare a fresh lid to be used during labeling: make two holes in the top of each lid but at opposite ends to inject and exhaust the labeling [14C]CO2. The inject hole should be at top near the rosettes and the exhaust hole should be at the bottom near the tips of the roots. The end of a paper clip, heated with a Bunsen burner, works well. With a syringe barrel filled with vacuum grease and without a needle, apply a bead of vacuum grease inside each lid to seal the labeling chambers. Use a small ball of modeling clay to cover the inject and exhaust holes (Figure 1B).
Note: (Optional) For improved [14C]CO2 circulation, a small blower fan, such as one typically used for cooling small electronic equipment (e.g., Pelonis Technologies Cat. No. RFB3004), can be oriented inside the labeling chamber to blow air across the plants. We typically use Scotch Removable Mounting Putty to hold the blower to the top or side of the chamber, with the wires emerging through the vacuum grease used to seal top and bottom halves of the chamber. Power is provided by D-cell batteries. Circulation is more important in larger chambers with more plants, and we do not use blowers in 100 x 100 mm plates.
Table 1. Schedule template to organize labeling and processing six chambers for long-distance phloem transport from source to sink organs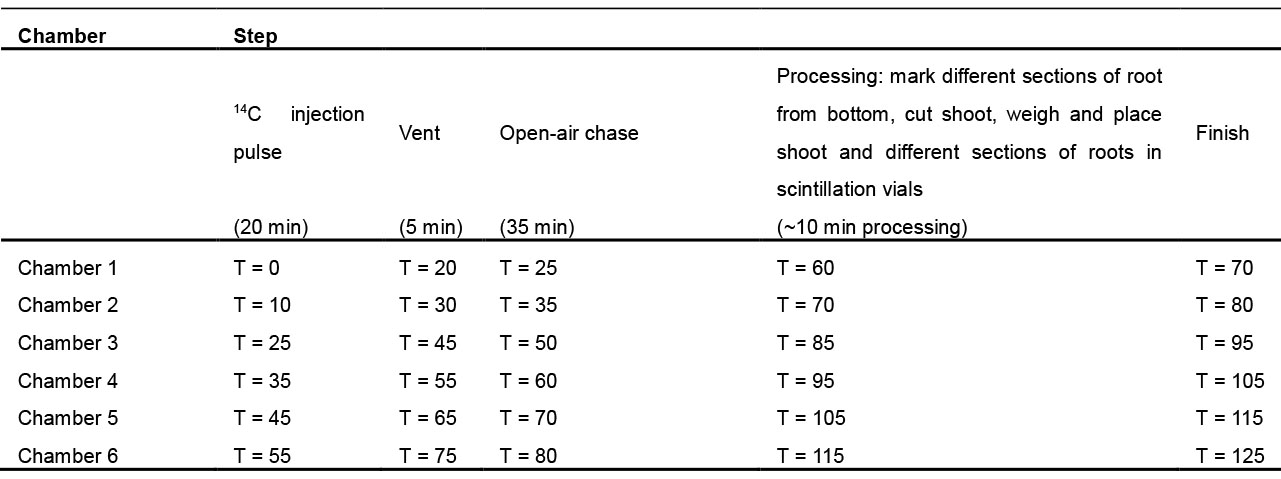
- Remove the lid of the first plate to be labeled and replace it with a lid equipped with inject and exhaust holes, and apply a bead of vacuum grease. Confirm by visual inspection that the vacuum grease seal is complete.
- To create [14C]CO2 for labeling, pipette 2.5 μl of [14C]NaHCO3 (2 μCi/μl, 2 mCi/ml) in a droplet near the syringe needle junction of a syringe barrel cut to ~3 cm. Place a 15 µl droplet of 80% lactic acid in the barrel, being careful to keep this droplet separate from the droplet of [14C]NaHCO3. Gently insert the plunger just inside the barrel (Figure 1C). Insert the needle through the injection hole of the culture plate to be labeled (remove the clay plug or push to the side), and arrange the modeling clay around it; a needle that is bent 45° works well (Figure 1D). The location of the injection hole and the size of the clay plug should not block light reaching the rosettes. Make sure the exhaust hole is also covered. Keep the droplets of [14C]NaHCO3 and lactic acid separate during these steps. Push the plunger gently to mix the lactic acid with [14C]NaHCO3 and release the [14C]CO2. Move the plunger back and forth to pump the [14C]CO2 gas into the labeling chamber; avoid injecting fluids into the chamber since the lactic acid can damage the plants. Remove the needle and cover the injection hole with modeling clay.
- Allow plants to do photosynthesis in the presence of [14C]CO2 for 20 min. This is the ‘pulse’ phase (Figure 1E).
- While plants in the first chamber are being labeled, replace the lid of the second culture dish with a pre-prepared lid with inject and exhaust holes and a vacuum grease bead, and make sure the seal is complete by visual observations (i.e., repeat Steps C3 and C4), and label as described in Step C5. Repeat for the third culture plate, etc.
- 20 min after injecting [14C]CO2, use the exhaust hole to vent the chamber through soda lime and capture unassimilated [14C]CO2 (Figure 1F). A column filled with soda lime and attached to a small air pump works well. After ~5 min of venting through soda lime, remove the lid used during labeling and replace with the original lid to allow gas exchange with unlabeled air. Do not try to reseal the lid with medical tape, but the plants should remain covered since they were grown in the high humidity of the culture dish. Allow photosynthesis to continue to provide 40 min of total ‘chase’ time.
Note: To ensure effective capture of unassimilated [14C]CO2 by the soda lime, it should be fresh and kept well-sealed between uses. Old soda lime should be discarded as 14C-labeled dry waste
Figure 1. Experimental procedure for labeling plants grown in culture plates with [14C]CO2. A. Arabidopsis plants on sterile ½ strength MS without sucrose solidified with 5 g/L gellan gum approximately 7 days after transfer from germination medium; the first three are one experimental line, the middle three are controls, and the last three are a second experimental line. Replicate plates will have a different order. Note that 1 cm of medium is removed and the plants were arranged so the shoots and hypocotyls are above the medium and the roots grow down the surface of the medium. The plate was grown in a vertical position at 15°. B. To convert the culture plate to a labeling chamber, a second lid with two holes for [14C]CO2 for injection (top) and exhaust (bottom) is prepared with a bead of vacuum grease to seal it with the bottom of the culture dish. C. A 1 ml syringe barrel cut to ~3 cm (cut end not shown) with a needle bent 45° and a small droplet of [14C]NaHCO3 (indicated by upper white arrow) and a larger droplet of 85% lactic acid (lower white arrow). The plunger is not yet inserted. D. Needle is inserted through the upper inject hole of the labeling chamber with the plunger inserted and the [14C]NaHCO3 and lactic acid mixed. Note that both the inject hole and the exhaust hole are sealed. E. Allow the plants to photosynthesize for 20 min. F. Remove the clay from both inject and exhaust holes and use Tygon tubing connected to a column of soda lime and a small air pump to exhaust the chamber for 5 min. G. Put the original plate back in place, but do not seal. Allow plants to do photosynthesis in regular air and transport photoassimilate for a 40 min ‘chase’ (5 min of exhausting the chamber and 35 min in regular air). H. Mark root length from root tip to shoot (e.g., 0.0-1.0 cm, 1.0-2.0 cm, etc.). I. Use a razor blade (shown) or fine scissors to cut the sections and transfer to separate scintillation vials containing 80% ethanol to stop metabolic reactions and extract metabolites. Alternatively, sections from each genotype can be pooled together into one scintillation vial for scintillation counting. Each plate should be considered a separate labeling experiment (i.e., one replicate), and the experimental samples should be standardized to controls before performing statistics on replicates.
- Photograph the plates to record growth on the day they are to be labeled. Turn on the 400 W metal halide light in the fume hood about an hour before labeling, so it reaches a stable intensity of ~130 μmol photons m-2 sec-1 at the working surface.
- Collection of tissues (Roots and Shoots) and scintillation counting
- Use fine scissors or a razor blade to cut the roots into sections as desired for the experiment. We usually cut the whole root from the shoot as a single heterotrophic organ, or we section the root into three regions: 0.0-1.0 cm from the tip (this is most actively growing and the strongest sink), 1.0-2.0 cm, and the remaining root (which will also include emerging lateral roots). Retain the shoot for scintillation counting.
- Collect the different sections of root tissues and the labeled shoot into 500 μl 80% ethanol in scintillation vials to terminate metabolism and extract the metabolites. The root sections and shoot of each plant can be collected and counted separately (preferred, but uses up a lot of scintillation vials) or tissues from each line in each plate can be pooled, and samples from each plate counted separately. Gently agitate the scintillation vials on a shaker for ~1 h to extract pigments and metabolites; greater volumes of 80% ethanol (~1 ml or more) may be required for larger rosettes. Add 500 μl commercial bleach to destroy pigments that may impede scintillation counting. Add sufficient scintillation fluid and mix thoroughly to get a clear, single-phase solution; 5-10 volumes of scintillation fluid to 1 volume of ethanol/bleach solution should suffice. Measure counts or disintegrations per minute in a scintillation counter.
- Use fine scissors or a razor blade to cut the roots into sections as desired for the experiment. We usually cut the whole root from the shoot as a single heterotrophic organ, or we section the root into three regions: 0.0-1.0 cm from the tip (this is most actively growing and the strongest sink), 1.0-2.0 cm, and the remaining root (which will also include emerging lateral roots). Retain the shoot for scintillation counting.
Data analysis
Each square culture plate constitutes an independent labeling experiment and photosynthetic incorporation of 14C[CO2] can vary between plates due to effects that are difficult to control. For example, since the plants in the plates are continuing to do photosynthesis while they are being prepared for labeling, the amount of atmospheric ‘cold’ CO2 in the vessel can vary from plate to plate when the 14C[CO2] is injected. This changes the 14C[CO2] specific activity during the pulse phase of the experiment. To control for this, control plants are included in each labeling chamber, and 14C[CO2] incorporation into experimental plants is expressed relative to these controls. That is, values obtained from experimental plants in each plate are standardized to a percent value of controls in the same chamber. Standardized values from separate plates are then combined as independent replicates. This removes the plate to plate variation in labeling efficiency to provide a more accurate representation of the differences in photoassimilation and transport between controls and experimental plants. Furthermore, to control for microenvironment effects in the plates, the order of control and experimental plants are altered among replicate chambers.
Recipes
- Half-strength Murashige and Skoog (MS) medium with sucrose for seed germination
Dissolve 2.22 g of MS into 800 ml of ddH2O
Add 10 g of sucrose, allow it to dissolve completely
Make the final volume 1,000 ml
Adjust the pH to 5.8 with 1 N KOH
Add 5 g gellan gum, mix the solution
Autoclave in liquid cycle for 20 min
Allow the medium to cool in a 55 °C water bath for 15-20 min
Pour into 100 x 25 mm circular Petri dishes - Half-strength Murashige and Skoog (MS) medium for experiments
Dissolve 2.22 g of MS into 1,000 ml of ddH2O
Adjust the pH to 5.8 with 1 N KOH
Add 5 g gellan gum, mix the solution
Autoclave in liquid cycle for 20 min
Allow the medium to cool in a 55 °C water bath for 15-20 min
Pour into 100 x 100 mm square plates
Acknowledgments
This protocol is based on methods published in (Dasgupta et al., 2014; Khadilkar et al., 2016). Work on phloem loading and long distance transport in B.G. Ayre’s laboratory is/was supported by the National Science Foundation 0344088, 0922546, 1121819, and 1558012. The authors report no conflicts of interest or competing interests.
References
- Ayre, B. G. (2011). Membrane-transport systems for sucrose in relation to whole-plant carbon partitioning. Mol Plant 4(3): 377-394.
- Clough, S. J. and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16(6): 735-743.
- Dasgupta, K., Khadilkar, A. S., Sulpice, R., Pant, B., Scheible, W. R., Fisahn, J., Stitt, M. and Ayre, B. G. (2014). Expression of sucrose transporter cDNAs specifically in companion cells enhances phloem loading and long-distance transport of sucrose but leads to an inhibition of growth and the perception of a phosphate limitation. Plant Physiol 165(2): 715-731.
- Khadilkar, A. S., Yadav, U. P., Salazar, C., Shulaev, V., Paez-Valencia, J., Pizzio, G. A., Gaxiola, R. A. and Ayre, B. G. (2016). Constitutive and companion cell-specific overexpression of AVP1, encoding a proton-pumping pyrophosphatase, enhances biomass accumulation, phloem loading, and long-distance transport. Plant Physiol 170(1): 401-414.
- Stadler, R., Lauterbach, C. and Sauer, N. (2005a). Cell-to-cell movement of green fluorescent protein reveals post-phloem transport in the outer integument and identifies symplastic domains in Arabidopsis seeds and embryos. Plant Physiol 139(2): 701-712.
- Stadler, R., Wright, K. M., Lauterbach, C., Amon, G., Gahrtz, M., Feuerstein, A., Oparka, K. J. and Sauer, N. (2005b). Expression of GFP-fusions in Arabidopsis companion cells reveals non-specific protein trafficking into sieve elements and identifies a novel post-phloem domain in roots. Plant J 41(2): 319-331.
- Yadav, U. P., Khadilkar, A. S., Shaikh, M. A., Turgeon, R. and Ayre, B. G. (2017a). Quantifying the capacity of phloem loading in leaf disks with [14C]sucrose. Bio Protoc 7(24): e2658.
- Yadav, U. P., Khadilkar, A. S., Shaikh, M. A., Turgeon, R. and Ayre, B. G. (2017b). Assessing rates of long-distance carbon transport in Arabidopsis by collecting phloem exudations into EDTA solutions after photosynthetic labeling with [14C]CO2. Bio Protoc 7(24): e2656.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Yadav, U. P., Khadilkar, A. S., Shaikh, M. A., Turgeon, R. and Ayre, B. G. (2017). Assessing Long-distance Transport from Photosynthetic Source Leaves to Heterotrophic Sink Organs with [14C]CO2. Bio-protocol 7(24): e2657. DOI: 10.21769/BioProtoc.2657.
- Khadilkar, A. S., Yadav, U. P., Salazar, C., Shulaev, V., Paez-Valencia, J., Pizzio, G. A., Gaxiola, R. A. and Ayre, B. G. (2016). Constitutive and companion cell-specific overexpression of AVP1, encoding a proton-pumping pyrophosphatase, enhances biomass accumulation, phloem loading, and long-distance transport. Plant Physiol 170(1): 401-414.
Category
Plant Science > Plant physiology > Nutrition
Plant Science > Plant physiology > Photosynthesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.