- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Genome Editing in Diatoms Using CRISPR-Cas to Induce Precise Bi-allelic Deletions
Published: Vol 7, Iss 23, Dec 5, 2017 DOI: 10.21769/BioProtoc.2625 Views: 15069
Reviewed by: Dennis NürnbergChristian SailerAgnieszka Zienkiewicz
Original research article
The authors used this protocol in:

Nov 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Genome editing in diatoms has recently been established for the model species Phaeodactylum tricornutum and Thalassiosira pseudonana. The present protocol, although developed for T. pseudonana, can be modified to edit any diatom genome as we utilize the flexible, modular Golden Gate cloning system. The main steps include how to design a construct using Golden Gate cloning for targeting two sites, allowing a precise deletion to be introduced into the target gene. The transformation protocol is explained, as are the methods for screening using band shift assay and/or restriction site loss.
Keywords: CRISPR-CasBackground
CRISPR-Cas is fast becoming a key method for molecular research. Based on a viral defence mechanism found in bacteria and archaea, CRISPR-Cas induces double-strand breaks (DSBs) at precise locations in the genome. It involves the use of a Cas9 nuclease which forms a complex with a chimeric single guide RNA (sgRNA) formed from CRISPR RNA (crRNA) and trans-activating crRNA (trRNA). Specificity is provided by a 20 nt sequence in the crRNA which corresponds with the target in the genome and guides Cas9 to the correct site by base complementarity. This means that the system is easily programmable and can be applied to new targets simply by changing the 20 nt sequence, provided that a protospacer adjacent motif (PAM) is present in the genome directly following the 20 nt target sequence. For the commonly used Cas9 isolated from Streptococcus pyogenes the PAM sequence is NGG. Gene editing is then achieved either by introducing mutations following imperfect repair by non-homologous end joining (NHEJ), cutting two sites and introducing a precise deletion or through homologous recombination. Since its application in the first eukaryotic systems (Cong et al., 2013; Mali et al., 2013), CRISPR-Cas has been used for genome editing in a wide range of organisms including two diatom species (Hopes et al., 2016; Nymark et al., 2016). Nymark et al. (2016) introduced mutations into the genome of Pheodactylum tricornutum using individual sgRNAs–the protocol for which can be found within Bio-protocol (Nymark et al., 2017). The protocol published in this paper focuses on gene editing in Thalassiosira pseudonana using two sgRNAs to introduce a precise deletion and allow simple screening using the band-shift assay previously described for identifying mutants in higher plants (Brooks et al., 2014). In addition, this method uses Golden Gate cloning (Weber et al., 2011; Engler et al., 2014)–a flexible modular system which allows sequences and cassettes to be easily interchanged and multiple modules to be assembled at once. Whilst this protocol describes targeting of two sites in the same gene to introduce a deletion, the construct can easily be altered to target different genes or greater numbers of genes as previously shown by Sakuma et al. (2014), who demonstrated knock-out of 7 genes using the Golden Gate cloning system.
Materials and Reagents
- 0.2 ml PCR tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: AB0620 )
- 10 µl filter pipette tips (STARLAB INTERNATIONAL, catalog number: S1121-3810 )
- 10 µl pipette tips (STARLAB INTERNATIONAL, catalog number: S1111-3810 )
- 200 µl pipette tips (STARLAB INTERNATIONAL, catalog number: S1111-0810 )
- 200 µl filter pipette tips (STARLAB INTERNATIONAL, catalog number: S1120-8810 )
- 1,000 µl pipette tips (STARLAB INTERNATIONAL, catalog number: S1111-6810 )
- 47 mm diameter 1.2 µm isopore filters (Merck, catalog number: RTTP04700 ) (optional)
- 1.5 ml Microcentrifuge tubes (Fisher Scientific, catalog number: 11926955 )
- 90 mm diameter Petri dishes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 263991 )
- 0.7 µm (M10) tungsten particles (Microcarriers) (Bio-Rad Laboratories, catalog number: 1652266 )
- Culture tubes (Fisher Scientific, catalog number: 11317824 )
- Macrocarriers (Bio-Rad Laboratories, catalog number: 1652335 )
- Macrocarrier holders (Bio-Rad Laboratories, catalog number: 1652322 )
- Stopping screens (Bio-Rad Laboratories, catalog number: 1652336 )
- 1,350 psi rupture discs (Bio-Rad Laboratories, catalog number: 1652330 )
- Thalassiosira pseudonana strain CCMP 1335 (Bigelow) https://ncma.bigelow.org/ccmp1335
- pCRTM8/GW/TOPOTM TA Cloning Kit with One ShotTM TOP10 E. coli (Thermo Fisher Scientific, InvitrogenTM, catalog number: K250020 )
- High efficiency competent E. coli (e.g., NEB 5-alpha Competent E. coli (High Efficiency)) (New England Biolabs, catalog number: C2987H )
- BsaI (New England Biolabs, catalog number: R0535S )
- BpiI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: ER1011 )
- Taq DNA polymerase (e.g., GoTaq Flexi DNA polymerase) (Promega, catalog number: M8291 )
- pICH47732:FCP:NAT (Addgene, catalog number: 85984 ) or pICH47732 (Addgene, catalog number: 48000 )
- pICH47742:FCP:Cas9YFP (Addgene, catalog number: 85986 ) or pICH47742 (Addgene, catalog number: 48001 )
- pICH47751 (Addgene, catalog number: 48002 )
- pICH47761 (Addgene, catalog number: 48003 )
- pICH41780 (Addgene, catalog number: 48019 )
- pAGM4723 (Addgene, catalog number: 48015 )
- pICH86966::AtU6p::sgRNA_PDS (Addgene, catalog number: 46966 )
- pCR8/GW:U6 (Addgene, catalog number: 85981 )
- Q5 Site-Directed Mutagenesis Kit (New England Biolabs, catalog number: E0554S )
- High fidelity DNA polymerase (e.g., Phusion High-Fidelity DNA Polymerase) (New England Biolabs, catalog number: M0530 )
- PCR clean up kit (e.g., Illustra GFX PCR DNA and Gel Band Purification Kit) (GE Healthcare, catalog number: 28-9034-70 )
- Plasmid mini prep kit (e.g., PureYield Plasmid Miniprep System) (Promega, catalog number: A1223 )
- Absolute ethanol (VWR, catalog number: 20821.330 )
- Agarose (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 17850 )
- Ethidium bromide for gel electrophoresis (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15585011 )
- TAE buffer for gel electrophoresis (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15558026 )
- T4 DNA ligase (Promega, catalog number: M1794 )
- 5-Bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-Gal) (Sigma-Aldrich, catalog number: B4252-50MG )
- IPTG (Sigma-Aldrich, catalog number: I6758-1G )
- Half salinity Aquil media (Price et al., 1989, full recipe at: https://ncma.bigelow.org/algal-recipes)
- Nourseothricin clonNAT (Werner BioAgents)
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: C5670-100G )
- Spermidine (Sigma-Aldrich, catalog number: S0266-1G )
- Isopropanol (VWR, catalog number: BDH1133-1LP )
- Compressed Helium supply
- Mix2seq kit (Eurofins)
- Tryptone (Sigma-Aldrich, catalog number: T7293 )
- Yeast extract (ForMedium, catalog number: YEA01 )
- Sodium chloride (Sigma-Aldrich, catalog number: S7653 )
- Agar (ForMedium, catalog number: AGA02 )
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A0166-5G )
- Spectinomycin dihydrochloride pentahydrate (Sigma-Aldrich, catalog number: S4014-5G )
- Carbenicillin disodium salt (Sigma-Aldrich, catalog number: C3416-250MG )
- Kanamycin sulphate (Sigma-Aldrich, catalog number: 60615-5G )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- Tris-HCl pH 8.0 (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15568025 )
- EDTA (Sigma-Aldrich, catalog number: EDS )
- LB media/LB agar (see Recipes)
- Lysis buffer (see Recipes)
Equipment
- Pipettes (Thermo Fisher Scientific, model: Finnpipette , volumes: 2 µl, 10 µl, 200 µl and 1,000 µl)
- NanoDrop ND-1000 (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 1000 )
- Laminar flow hood (Walker safety cabinets, model: Class II 1290 Recirc Gen 6 )
- Benchtop microfuge (Eppendorf, model: 5418 R )
- Centrifuge (Hettich Lab Technology, model: ROTINA 380 R )
- Autoclave (Prestige Medical, catalog number: 210004 )
- Tweezers
- Vortex (Mo Bio Laboratories, model: Vortex Genie® 2 )
- Shaking incubator (37 °C) (IKA, model: KS 4000 i control )
- Light incubator at 20 °C (Sanyo versatile Environmental Test chamber)
- PCR thermocycler (Bio-Rad Laboratories, model: T100TM Thermal cycler )
- Light Microscope and Neubauer chamber (VWR, catalog number: 631-0696 ) or Coulter Counter (Beckman, model: Multisizer 3 ) for counting cells
- PDS-1000/He biolistic microparticle delivery system (particle gun) (Bio-Rad Laboratories, catalog number: 1652257 )
- Vacuum pump for cell filtration (Welch Vacuum, model: 2534C-02 ) (optional)
- Nalgene filtration funnel (Thermo Fisher Scientific, Thermo ScientificTM, model: DS0320-5045 ) (optional)
- High Vacuum pump for microparticle delivery system (Uniweld Products, model: HUMM•VACTM HVP6 )
- Gel electrophoresis tank (Fisher Scientific, FisherbrandTM, model: Midi Plus Horizontal Gel System )
- Electrophoresis power supply (Consort, model: EV243 )
Procedure
- Designing sgRNAs
sgRNAs are designed to facilitate screening once mutations have been introduced. This protocol describes two screening methods: the band shift assay and restriction site loss. We have found the band shift assay to be the most effective–this method is based on introducing a deletion using two sgRNAs which can be screened by amplifying the target and looking for a shorter band on an agarose gel. It has the added benefit of introducing very precise and large deletions. This can be used as a stand-alone method without the need for restriction site loss. Restriction site loss is slightly more complicated and limits the sgRNA design as DSBs need to coincide with a restriction recognition site (preferably at least a 5 nt site cutter). When mutations are introduced through CRISPR-Cas, the restriction site is no longer active and the restriction enzyme cannot cut. This method has been included as it can be a useful way to enrich mutated targets or screen if only one sgRNA is functional.- This protocol uses the chimeric sgRNA for use with the S. pyogenes Cas9 with the following sequence:NNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT
- The underlined sequence shows the programmable region which corresponds to the 20 nt target sequence in the genome.
- In the genome, this sequence is immediately followed by the NGG PAM. Therefore targets need to be identified in the genome with the sequence N20NGG.
Note: The target sequence of the sgRNA itself should only contain the N20 sequence–i.e., it should not contain the PAM. - In addition, a U6 promoter which recruits RNA polymerase III (pol III) is used to drive expression of the small non-coding sgRNAs. This means that the sgRNA needs to start with a ‘G’ to activate transcription. This can either be incorporated into the target design meaning that targets with the sequence GN19NGG need to be identified OR an additional G can be added to the 5’ end of the sgRNA.
- The string of 6 Ts in bold acts as a terminator for transcription when expressing the sgRNA using pol III.
- The underlined sequence shows the programmable region which corresponds to the 20 nt target sequence in the genome.
- Two tools are used to design sgRNAs targets:
- RGEN Cas-Designer: http://www.rgenome.net/
- Includes an off-target finder for several algal and diatom genomes including T. pseudonana. This allows the researcher to reject targets if similar sequences occur elsewhere in the genome which may also be cut.
- Allows specified sequences to be omitted such as introns.
- Can search for targets with several different PAMs if the researcher wishes to change the Cas9.
- Includes an off-target finder for several algal and diatom genomes including T. pseudonana. This allows the researcher to reject targets if similar sequences occur elsewhere in the genome which may also be cut.
- Broad Institute sgRNA designer: https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design
This is used to give on-target scores. The algorithm used by this program is based on empirically tested sgRNAs and predicts their cutting efficiency. Try to choose targets which give a cutting efficiency as close to 1 as possible.
- RGEN Cas-Designer: http://www.rgenome.net/
- Design two sgRNAs with cut sites about 50-100 bp apart
- A deletion of 50-100 bp gives a clear difference between edited targets and the WT (Belhaj et al., 2013; Brooks et al., 2014).
Note: A larger deletion can be chosen if necessary–research suggests that mutations from larger deletions occur less frequently, however, deletions up to 10 kb are still efficient (Zheng et al., 2014). - Either target the active site of the gene or the beginning of the gene if a frame shift is expected.
Note: Whole genes can also be removed by targeting the ends of the genes or the end of the promoter/start of the terminator. This can be useful if the active site is unknown or there are problems with designing sgRNAs, for example, when a large number of repeat sequences are present.
- A deletion of 50-100 bp gives a clear difference between edited targets and the WT (Belhaj et al., 2013; Brooks et al., 2014).
- If the restriction site loss method is being used for screening, targets need to be designed so that Cas9 cut sites sit over a restriction recognition site or within the deletion site.
Targets that position cut sites over restriction sites can be determined using software such as PhytoCRISPRex (Rastogi et al., 2016) or by running the target gene through a restriction finder such as the Emboss restriction tool (http://emboss.bioinformatics.nl) and cross-referencing location of the restriction sites with the position of the Cas9 cuts sites/deletions.
Note: It may not always be possible to design sgRNAs exactly within these specifications, for example, sgRNAs with lower on-target scores may need to be used due to positioning constraints (we have successfully used sgRNAs with a score of 0.4), or a slightly smaller deletion may be necessary. Bear in mind that even if it is possible, targets still need to be empirically tested as other factors such as accessibility may affect their activity. As a result, it is recommended to design at least two sets of sgRNAs for each target gene/sequence.
- This protocol uses the chimeric sgRNA for use with the S. pyogenes Cas9 with the following sequence:
- Making the construct
The construct is assembled using the Golden Gate system which builds the plasmid in modules using pre-set backbones and pre-set overhangs between sequences (Weber et al., 2011). The basis behind Golden Gate cloning is the use of restriction enzymes BsaI and BpiI, which cut outside of the restriction recognition sites, allowing specific 4 nt overhangs to be created. This allows fragments to be joined together in a specific order and within a single reaction for each level. As the restriction recognition site and cut site are separate, restriction digest and ligation of the insert leads to complete removal of the site, meaning that once the correct assembly is achieved the construct can no longer be cut. This is carried out over three levels, L0, L1 and L2.
L0 modules contain sequences such as promoters, coding regions and terminators. These are assembled together to form cassettes in L1 backbones. L1 modules are then assembled into the final L2 backbone to create a construct with all the necessary sequences required for CRISPR-Cas. This section describes the construct assembly for CRISPR-Cas in T. pseudonana for targeting two sites in the genome, however, it can easily be adapted for use with other organisms, different modules (e.g., different Cas9, promoters or selective markers) or to target more than two sites. The latter makes this cloning approach ideal for targeting multiple genes.
L1 backbones contain BsaI sites to allow cloning of L0 modules, as well as BpiI sites to allow L1 modules to be cloned into the L2 backbone. L0 modules are created by amplifying the desired fragments with Taq DNA polymerase and cloning into a pCRTM8/GW/TOPO vector. In order to clone L0 modules into L1 backbones, BsaI sites need to be included through the forward and reverse primers when amplifying the desired fragment. In addition, the overhangs which are created when the BsaI sites are digested need to be the same between adjacent modules. All L1 backbones used within this protocol give standardised overhangs of GGAG and GCTT when digested with BsaI. As a result, the first L0 module in the sequence to be cloned in must contain a GGAG overhang at the 5’ end and the last module must contain a GCTT overhang at the 3’ end. Internal overhangs between L0 modules are decided by the researcher and can be designed to give a seamless join. Below is an example of an amplicon for cloning into L0 showing the start and end of the sequence:
TggtctcaggagAGCTTGCGCTTTTTCCGAG…CTGATTTACCAAACCAATACCAAAatgtgagacct
Bold letters denote the BsaI site, underlined regions show the 4 nt overhang generated by restriction with BsaI and the italicised region shows the fucoxanthin chlorophyll a/c-binding protein (FCP) promoter sequence. The first overhang corresponds with the overhang for the end of the L1 backbone. The second overhang corresponds with the overhang from the adjacent Cas9 module. This has been designed so that the overhang contains the ATG at the start of the Cas9, giving a seamless join.
Figure 1 gives a general overview of the assembly process used in this protocol.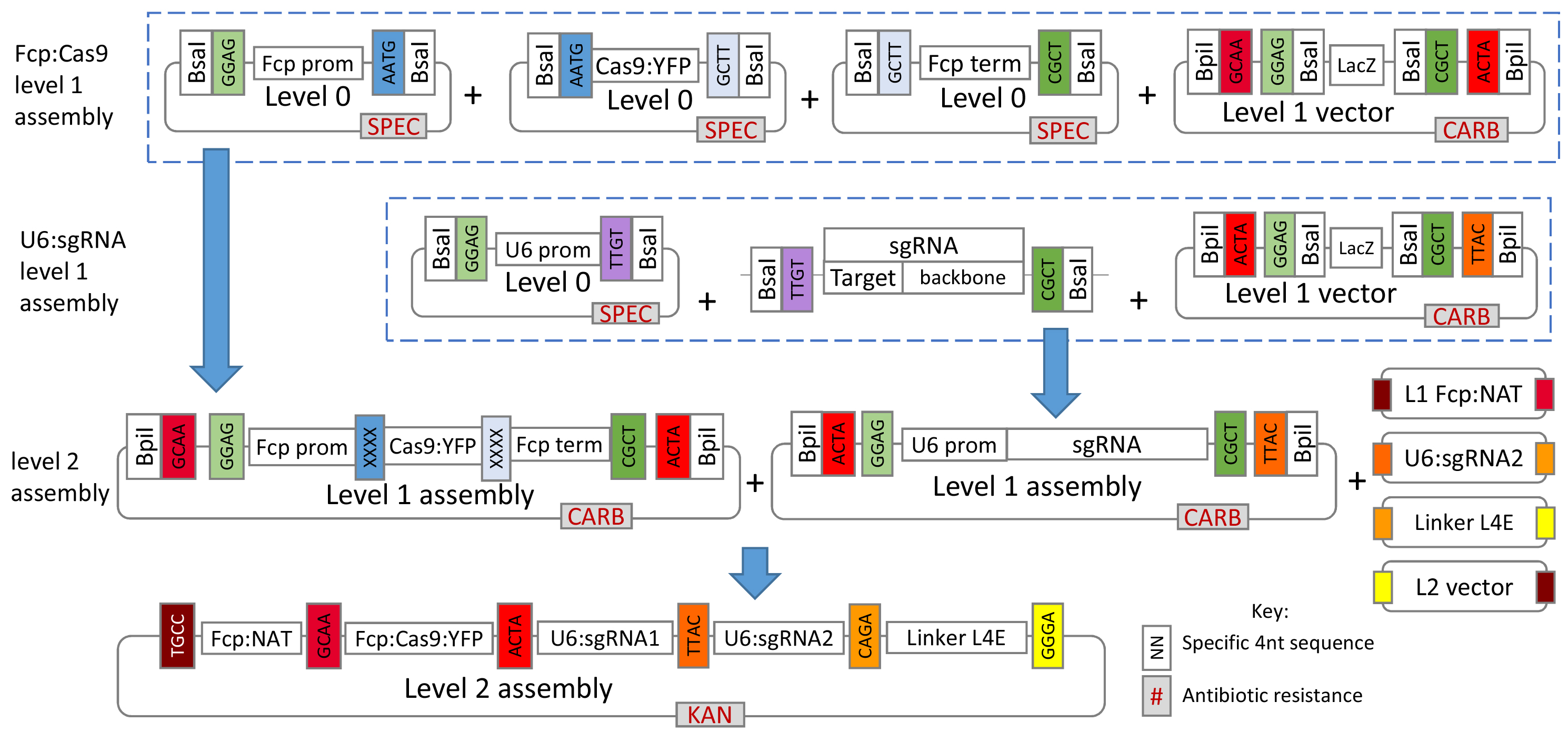
Figure 1. Overview of the Golden Gate cloning system for assembly of the CRISPR-Cas construct. For T. pseudonana, plasmids for the L0 U6 promoter, L1 FCP:NAT cassette and L1 FCP:Cas9:YFP cassette are already available from Addgene (see the link below). For additional L0 T. pseudonana modules please contact the authors.- The protocol describes creation and assembly of all modules so that researchers can modify the overall system if desired, however, several of the L0 and L1 modules described are already available from Addgene at https://www.addgene.org/Thomas_Mock/. In addition, modules for P. tricornutum will soon be available from Addgene on the same website.
- Remove BsaI and BpiI sites from inserts
- The original pTpFCP:NAT (Poulsen et al., 2006) cassette for nourseothricin resistance contains a BsaI site in the FCP promoter and a BpiI site in the NAT gene. These sites need to be removed by site-directed mutagenesis to prevent unwanted restriction digest during Golden Gate cloning.
- Using pTpFCP/NAT (Poulsen et al., 2006) as a template, remove the BsaI site using forward primer: TCCGCGGCAGaTCTCTGTCG, reverse primer: AGAAGTACCGTGTTGTTGCAGTG, and a Q5 site-directed mutagenesis (SDM) kit (NEB). Repeat SDM on the BpiI site in the NAT gene using the resulting pTpFCP/NAT plasmid (containing the domesticated FCP promoter) as a template, forward (F) primer: CGACACCGTaTTCCGCGTCAC and reverse (R) primer: GTGGTGAAGGACCCATCCAG.
Note: If using the TpFCP/NAT cassette there is no need to domesticate this sequence as the L1 pICH47732:FCP:NAT module is already available from Addgene. - If using different sequences than those described in this protocol, remove BpiI and BsaI sites in a similar manner as described above. Software for designing SDM primers is available from NEB alongside the kit (http://nebasechanger.neb.com/).
- The original pTpFCP:NAT (Poulsen et al., 2006) cassette for nourseothricin resistance contains a BsaI site in the FCP promoter and a BpiI site in the NAT gene. These sites need to be removed by site-directed mutagenesis to prevent unwanted restriction digest during Golden Gate cloning.
- Using U6 promoters in diatoms
- All known U6 promoters for diatoms are shown in Table 1. The U6 promoter for P. tricornutum was first described by Nymark et al. (2016) and activity of the T. pseudonana (Hopes et al., 2016) and Fragilariopsis cylindrus promoters (unpublished) have been determined in our lab. In addition, the exact end of the U6 promoters for T. pseudonana and F. cylindrus have been determined empirically using the 5’ TSO RACE (Pinto and Lindblad, 2010) method.
Table 1. U6 promoters in diatoms and the overhangs required for scar less assembly with the sgRNA. The TATA box is highlighted in yellow and the overhang (last 4 nt of the promoter) in green.
- When cloning the U6:sgRNA cassette through Golden Gate, it is important to ensure a scarless junction between the end of the U6 promoter and the start of the sgRNA so that extra nucleotides are not transcribed at the 5’ end of the sgRNA. This is achieved by designing the 4 nt overhang between the U6 promoter and sgRNA as the last 4 nt of the U6 promoter. The overhang necessary to create a scarless junction between the U6 promoter and sgRNA varies for each U6 promoter and requires the overhang to be built into the reverse primer for amplifying the U6 promoter and the forward primer for amplifying the sgRNA. These primers can be found in Table 2 below:
Table 2. Primers used to introduce the overhang between diatom U6 promoters and sgRNAs. BsaI sites are underlined and overhangs highlighted in green. Ns denote the target site. Uppercase letters A, C, G or T show the section of the primer that will anneal with the initial template.
- All known U6 promoters for diatoms are shown in Table 1. The U6 promoter for P. tricornutum was first described by Nymark et al. (2016) and activity of the T. pseudonana (Hopes et al., 2016) and Fragilariopsis cylindrus promoters (unpublished) have been determined in our lab. In addition, the exact end of the U6 promoters for T. pseudonana and F. cylindrus have been determined empirically using the 5’ TSO RACE (Pinto and Lindblad, 2010) method.
- Creating L0 modules
- A full list of primers, annealing temperatures and extension times can be found for all Golden Gate PCR reactions in Table 3.
Table 3. PCR primers for Golden Gate cloning. BsaI sites are underlined. Overhangs are shown in bold. F denotes the forward primer and R the reverse primer. The string of Ns in the forward sgRNA primer needs to be replaced by the 20 nt target sequence designed for each gene/target. Uppercase letters A, C, G or T show the section of the primer that will anneal with the initial template.
- L0 modules contain a Sm/SpR gene to allow selection by spectinomycin in E. coli.
- Amplify the FCP promoter from the domesticated pTpFCP/NAT cassette using F primer: tggtctcaggagAGCTTGCGCTTTTTCCGAG and R primer aggtctcacatTTTGGTATTGGT TTGGTAAATCAG (Table 3, numbers 1 and 2) using Taq DNA polymerase. Clone the PCR product directly into a pCRTM8/GW/TOPO vector.
- Amplify Cas9:YFP using F primer: aggtctcaaATGGACAAGAAGTACTCCATTGG and R primer: aggtctcaaagcTCACTTGTACAGCTCGTCCATG (Table 3, numbers 3 and 4) using high fidelity (HF) Phusion polymerase. Clean the reaction with a PCR purification kit and incubate with Taq polymerase (Table 4) for 20 min at 72 °C before cloning into the pCRTM8/GW/TOPO vector.
Note: Amplification of the Cas9 is carried out with HF polymerase to reduce the chance of errors during amplification since Cas9:YFP is a large sequence of 4.8 kbp. As HF polymerases do not add a 3’ ‘A’ overhang, a separate incubation with Taq is required.
Table 4. Reagents for Cas9:YFP PCR incubation with Taq to add ‘A’ overhangs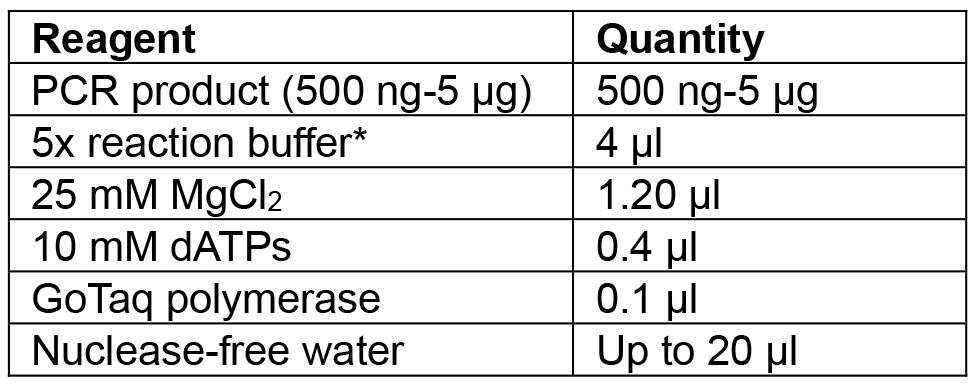
- Amplify the FCP terminator from domesticated pTpFCP/NAT using F primer: aggtctcagcttATACTGGATTGGTGAATCAATG and R primer: tggtctcaagcgGAGAA CTGGAGCAGCTAC (Table 3, numbers 5 and 6) using Taq DNA polymerase. Clone the PCR product directly into a pCRTM8/GW/TOPO vector.
- Amplify the U6 promoter from T. pseudonana genomic DNA using the F primer: cggtctcaggagCTTCATCAAGAGAGCAACCA and R primer: aggtctcaACAATTTCGG CAAAACGT (Table 3, numbers 7 and 8) using Taq DNA polymerase. Clone the PCR product directly into a pCRTM8/GW/TOPO vector. See section 3 for using U6 promoters in diatoms. This module is already available from Addgene.
- Due to the guide sequence changing between targets, sgRNAs are not cloned into L0 vectors, instead they are directly assembled into the L1 vector as a PCR product. Target sequences are introduced through the F primer when amplifying the scaffold. Amplify the scaffold from pICH86966::AtU6p::sgRNA_PDS using F primer: aggtctcattgtNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAG and R primer: tggtctcaagcgTAATGCCAACTTTGTACAAG (Table 3, numbers 11 and 12). It is fine to use either Taq or HF DNA polymerase. The run of Ns in the forward primer represents the target sequence. Following amplification, PCR products are purified using a PCR clean-up kit.
- A full list of primers, annealing temperatures and extension times can be found for all Golden Gate PCR reactions in Table 3.
- Screening L0 modules
- Screen all L0 modules before assembly into L1 vectors. Perform colony PCR using the same primers used to amplify fragments for cloning into L0 modules (Table 3) or digest the L0 module to check for size. Sequence L0 modules that contain inserts of the correct size using primers provided in the pCRTM8/GW/TOPO TA kit.
- Colony PCR:
- Prepare Master Mix for the number of colonies to be tested according to Table 5 and aliquot 20 µl into PCR tubes.
Table 5. Reagents for colony PCR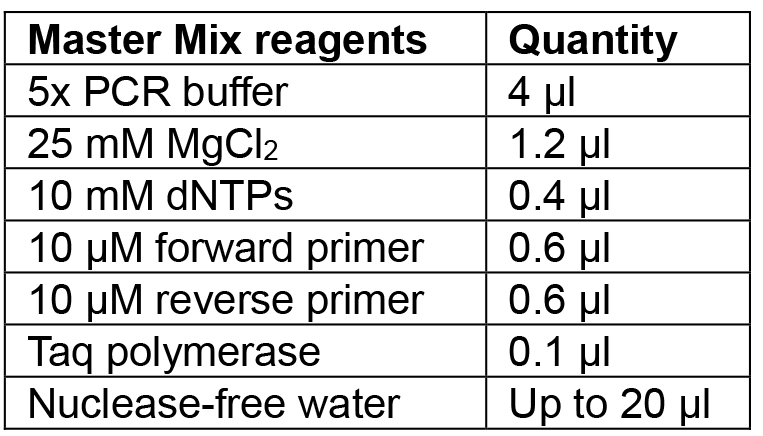
- Lightly touch a 10 µl pipette tip to each colony (label the colony so that it can be picked following screening), mix using the pipette and briefly spin down.
- Run the following PCR cycle.
1) Initial denaturation: 95 °C for 2 min
2) 35 cycles:
Denature at 95 °C for 30 sec
Anneal for 30 sec (see Table 3 for annealing temperatures)
Extend at 72 °C (see Table 3 for annealing times)
3) Carry out the final extension at 72 °C for 10 min
- Prepare Master Mix for the number of colonies to be tested according to Table 5 and aliquot 20 µl into PCR tubes.
- Restriction digest:
Extract plasmids using a plasmid mini-prep kit, measure the DNA concentration and digest with BsaI to check the size of the insert.
Note: A NanoDrop spectrophotometer is used to measure DNA concentration.
- Screen all L0 modules before assembly into L1 vectors. Perform colony PCR using the same primers used to amplify fragments for cloning into L0 modules (Table 3) or digest the L0 module to check for size. Sequence L0 modules that contain inserts of the correct size using primers provided in the pCRTM8/GW/TOPO TA kit.
- L1 assembly
- Four L1 modules are assembled
pICH47732:FCP:NAT (available from Addgene)
pICH47742:FCP:Cas9YFP (available from Addgene)
pICH47751:U6:sgRNA1
pICH47761:U6:sgRNA2 - Golden Gate assembly is carried out in a single reaction for each L1 assembly.
- Set up the reaction as shown in Table 6.
Table 6. Reagents for L1 Golden Gate assembly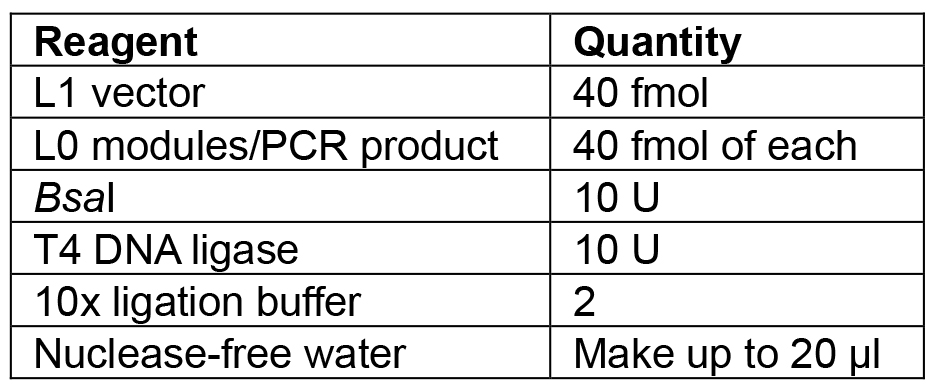
- Incubate under the following conditions
37 °C for 5 h
50 °C for 5 min
80 °C for 10 min - Transform 5 µl of the reaction into 50 µl of competent E. coli.
- L1 modules contain the cassette for carbenicillin resistance for selection in E. coli.
Note: Insertion of the correct insert should remove the LacZ gene for blue to white colony screening when incubated with X-gal and IPTG.
- Set up the reaction as shown in Table 6.
- Amplify the FCP:NAT cassette from domesticated pTpFCP:NAT with forward primer: tggtctcaggagCTCGAGGTCGACGGTATC and reverse primer aggtctcaagcgCGCAATTA ACCCTCACTAAAGG (Table 3, numbers 9 and 10) using HF Phusion DNA polymerase. Clone into L1 backbone pICH47732. This module is already available from Addgene.
- Clone the L0 FCP promoter, L0 Cas9:YFP and L0 FCP terminator into L1 backbone pICH47742. This module is already available from Addgene.
- Clone the L0 U6 promoter and sgRNA1 PCR product into L1 backbone pICH47751
- Clone the L0 U6 promoter and sgRNA2 PCR product into L1 backbone pICH47761
- Four L1 modules are assembled
- Screen L1 assemblies
Screen L1 assemblies either by colony PCR as described or restriction digest with XbaI or EcoRV. Sequence inserts using primers: CCCACTCTGTGAAGACAA and GCCAATATATCCTGTCAAACAC which anneal just upstream and downstream of the insert sites respectively. - L2 assembly
- Assemble L1 modules into the Level 2 backbone along with the linker pICH41780 which joins the 4th module to the vector.
Note: If a different number of modules are to be assembled, the appropriate linker needs to be used to ensure the overhang between the last module and the linker is correct. Details of further Golden Gate modules can be found in Engler et al. (2014). - Set up the reaction as shown in Table 7.
Table 7. Reagents for L1 Golden Gate assembly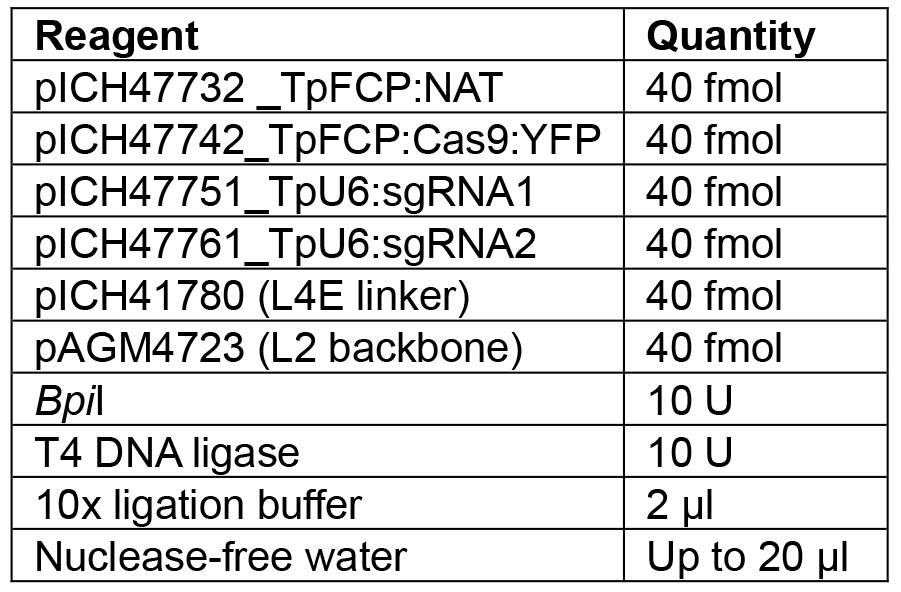
- Incubate under the following conditions
37 °C for 5 h
50 °C for 5 min
80 °C for 10 min - Transform 5 µl of the reaction into 50 µl of competent E. coli and prepare plasmids using a plasmid mini-prep kit.
- L2 modules contain the cassette for kanamycin resistance for selection in E. coli.
Note: Insertion of the correct inserts should result in removal of a gene for canthaxanthin synthesis resulting in a colour change from orange/pink to white.
- Assemble L1 modules into the Level 2 backbone along with the linker pICH41780 which joins the 4th module to the vector.
- Screen L2 assembly
- Check the plasmid is the correct size by digesting with XbaI or EcoRV and running on an agarose gel.
Note: Restriction sites will vary depending on the construct–choose enzymes that will linearize the plasmid or give a banding pattern that is distinct from the original L2 backbone. - Sequence using the primers shown in Table 8.
Table 8. Primers for sequencing L2 constructs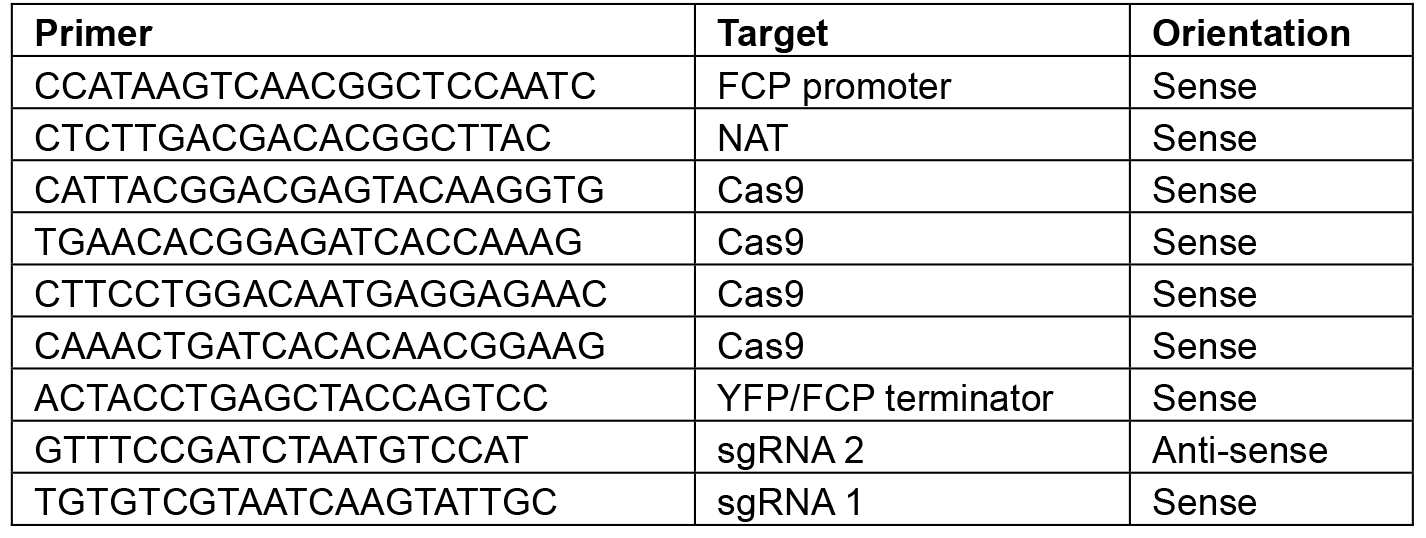
- Once plasmids have been successfully screened, perform a plasmid maxi-prep kit to produce enough plasmid for transformation.
- Ethanol precipitate plasmids to remove any traces of salt and dilute to 1 µg/µl.
- Check the plasmid is the correct size by digesting with XbaI or EcoRV and running on an agarose gel.
- Transform the construct into T. pseudonana
- Transformation of the CRISPR-Cas construct into T. pseudonana follows the T. pseudonana protocol set out by Poulsen et al. (2006) and the P. tricornutum transformation protocol from Kroth (2007). For ease of use, a combination of both protocols is included in this paper.
- Where possible all steps should be prepared aseptically under a laminar flow hood.
- Grow T. pseudonana strain CCMP 1335 to exponential phase (approximately 1 x 106 cells/ml) in 24 h light (100-140 μE) at 20 °C in half salinity media (all salt concentrations are halved compared to the full salinity Aquil media recipe, but nutrients, vitamin and trace metal concentrations remain the same). Algal media recipes can be found in full on the Bigalow website (https://ncma.bigelow.org/algal-recipes).
Note: Full salinity media such as Aquil (Price et al., 1989) can be used to culture T. pseudonana but cells grow just as well in half salinity and the salt concentration needs to be reduced for effective nourseothricin activity. - Prepare tungsten particles
- The following preparation uses 60 mg of tungsten particles but can be scaled down.
- Weigh 60 mg of 0.7 µm tungsten particles.
- Wash by adding 1 ml of 100% ethanol and vortex for 3 min.
- Centrifuge at 13,000 x g for 1 min and remove supernatant.
- Add 1 ml of sterile, nuclease-free water and mix. Re-centrifuge and remove supernatant.
- Repeat the above step two additional times.
- Resuspend the particles in 1 ml of water and aliquot 50 µl into tubes.
- Store at -20 °C for up to two weeks or keep on ice until needed.
- The following preparation uses 60 mg of tungsten particles but can be scaled down.
- Prepare plates for shooting and selection
- Plates for shooting are made with 1.5% agar and half salinity media.
- Plates for selection are made with 0.8% agar, half salinity media and 100 µg/ml nourseothricin.
- When making plates, in order to avoid precipitation, separately make up and autoclave 2x concentrated agar solution and 2x concentrated media (or in this case full salinity, as the final media needs to be half salinity), without nutrients, vitamins, trace metals or antibiotics. Once solutions have cooled to 50 °C, mix and add nutrients, vitamins, trace metals and, in the case of the selective plates, antibiotics. Plates can then be poured.
- Plates for shooting are made with 1.5% agar and half salinity media.
- Prepare cells
- 5 x 107 cells are used per shot and shots are performed in triplicate. A positive control and negative control are also carried out. As a result transformation of one construct plus controls requires enough culture for 9 shots = 4.5 x 108 cells.
- Either, gently spin cells down in a centrifuge at 3,000 x g for 10 min and discard all the supernatant. Resuspend in 100 µl of media per shot. 100 µl of suspension is then spread in a 5 cm diameter circle in the centre of each 1.5% agar plate, and allowed to dry at room temperature.
- Or, filter 5 x 107 cells onto 47 mm diameter, 1.2 µm isopore filters using gentle vacuum filtration. Place the filter onto a 1.5% agar plate.
- 5 x 107 cells are used per shot and shots are performed in triplicate. A positive control and negative control are also carried out. As a result transformation of one construct plus controls requires enough culture for 9 shots = 4.5 x 108 cells.
- Prepare particles
- Add the following in order to a 1.5 ml microcentrifuge tube:
1) 50 µl of prepared tungsten particles.
2) 5 µg of the plasmid in 5-10 µl of water (for the negative control just use water).
3) 50 µl of 2.5 M CaCl2 (store solution at -20 °C).
4) 20 µl of 0.1 M spermidine (store solution at -20 °C for up to one month). - Vortex the tube for 1 min.
- Briefly spin down the particles in a centrifuge and remove the supernatant.
- Add 250 µl of 100% ethanol and vortex until homogenous.
- Briefly spin down the particles in a centrifuge and remove the supernatant.
- Resuspend in 50 µl of 100% ethanol. Store on ice. Particles need to be used within 1 h of preparation.
- Add the following in order to a 1.5 ml microcentrifuge tube:
- Microparticle bombardment
- Set up the particle gun according to manufacturer’s instructions.
- Clean the particle gun by wiping with 70% ethanol.
- Autoclave the rupture disc retaining cap, microcarrier launch assembly, macrocarrier holders, stopping screes and tweezers.
- Sterilise 1,350 psi rupture discs by dipping in isopropanol and allow to dry.
- Sterilise macrocarriers by dipping in 70% ethanol and allowing to dry.
- Before shooting cells, the helium lines need to be cleared. This can be carried out by performing the following steps required for shooting but without using cells or particles.
- Place the macrocarriers inside the macrocarrier holders.
- Vortex the coated tungsten particles, and keeping the particles mixed, pipette 10 µl onto the centre of the macrocarrier. Temporarily turn off the laminar flow hood whilst drying particles to prevent loss.
- Place a 1,350 psi rupture disc into the retaining cap, and screw the cap tightly into the assembly using the provided tool.
- Once the particles have dried, load the macrocarrier and stopping screen into the launch assembly. The launch assembly is then placed in the biolistic chamber.
- Place a plate with cells at a flight distance of 7 cm in the biolistic chamber.
- Close and turn on the vacuum pump until a vacuum of 25 Hg is reached.
- Fire particles into the cells.
- Following particle bombardment rinse cells from each plate into 25 ml of media without antibiotics and incubate for 24 h under standard growth conditions.
- Following the 24 h incubation, count cells and plate 5 lots of 5 x 106 cells from each transformation onto selective plates.
- Using the cells from the negative control, also spread cells onto plates without antibiotics as a positive growth control.
Note: T. pseudonana requires relatively small concentrations of cells to be plated out in order for nourseothricin to be effective compared to other diatom transformation systems such as P. tricornutum. As a result, there will be a large quantity of transformed cells left in media after plating. If desired, additional plates can be spread, or liquid cultures can be maintained until colonies start to appear on plates in case further plates are needed.
- Set up the particle gun according to manufacturer’s instructions.
- Transformation of the CRISPR-Cas construct into T. pseudonana follows the T. pseudonana protocol set out by Poulsen et al. (2006) and the P. tricornutum transformation protocol from Kroth (2007). For ease of use, a combination of both protocols is included in this paper.
- Screen transformants by PCR/Band shift assay
- Once colonies appear (after approximately 10 days), transfer into selective liquid media.
- Resuspend a colony in 20 µl of media and transfer 10 µl to a PCR tube for colony PCR. The remaining 10 µl can be used to grow up individual cell lines.
Place the remaining 10 µl in 1 ml of selective liquid media. Once cells reach around 1 x 106 cells/ml, this can then be used to inoculate larger volumes. - Spin the 10 µl of cells, set aside for colony PCR, down in a centrifuge for 1-2 min at full speed and remove the supernatant.
- Resuspend cells in lysis buffer (see Recipes).
- Incubate on ice for 15 min.
- Incubate at 95 °C for 10 min.
- Use 1 µl as template in a 20 µl PCR reaction using Taq polymerase.
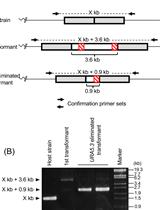
- Amplify the target region. For the urease gene described in Hopes et al. (2016) the forward primer: AAACAGACCACCTTCACCTC and reverse primer: CTCCACCTGTACGTCTCG were used. Run PCR products on a 1% w/v agarose gel at 4-5 V/cm (80-100 V with the specified gel system). In the Hopes et al. (2016) paper, as the deletion was only 37 nt, instead of the 1% w/v gel, a 2% w/v agarose gel was run at 3 V/cm (60 V with the specified gel system) to visualise the shift in band size compared to the WT. See Figure 2 for gels demonstrating deletion induced band shifts.
- Amplify a section of the Cas9 gene using forward primer: CCGAGACAAGCAGAGTGGAAAG and reverse primer: AGAGCCGATTGATGTCCAGTTC. Run PCR products on a 1% w/v agarose gel at 4-5 V/cm (80-100 V with the specified gel system) to check for the presence of Cas9.
Note: We found that around 11% of primary clones contained Cas9 but 100% of clones with Cas9 contained a deletion. Screen at least 30-40 colonies to isolate several primary clones with deletions. - Include WT samples and negative controls using water instead of template in all PCR reactions.
- Due to the nature of CRISPR-Cas, primary colonies can be mosaic with a mixture of WT and mutant cells (Figure 2). In order to procure a clean cell-line with one clone, cultures from primary clones, with evidence of mutations, need to be re-spread onto additional selective plates. Streak 100 µl of cells from the primary cultures at exponential phase onto new selective plates and incubate until colonies appear. Sub-clones can then be screened as shown above.
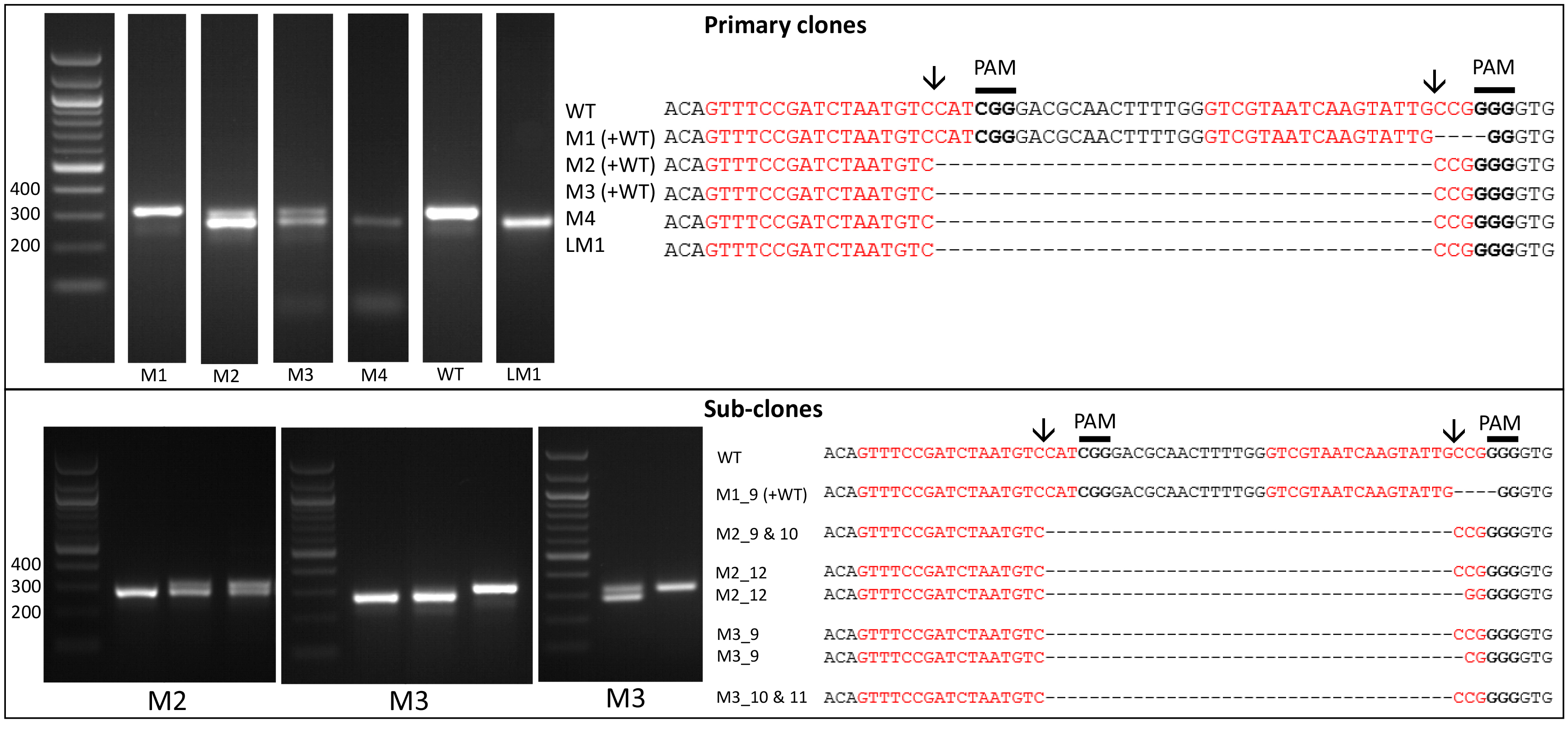
Figure 2. Screening of the CRISPR-Cas induced genome editing in T. pseudonana. Screening by PCR and sequencing. Expected sgRNA cut indicated by ↓. Red text shows the sgRNA target sequence and bold text the PAM motif. Primary clones: Several primary clones contain sequences showing both CRISPR-induced mutations and the wildtype (WT) sequence, as seen by the presence of two bands following PCR, these are indicated by (+WT). Remaining samples represent different cell lines. M1 shows a 4 nt deletion from the second sgRNA whilst mutants M2-M4 show a 37 nt deletion between the two CRISPR-Cas cut sites. Sub-clones: The gel shows examples of a selection of sub-clones derived from the primary clones. Sub-clones are labelled according to the primary clone and sub-clone number. With the exception of M1_9, which gives a WT sequence and 4 nt deletion as seen in the primary clone, all sub-clones chosen for sequencing are bi-allelic. Two-thirds of sequenced bi-allelic sub-clones show a single sequence with a 37 nt deletion suggesting that both alleles carry the same mutation. In sub-clones where mutations differ between alleles both sequences are shown.
- Once colonies appear (after approximately 10 days), transfer into selective liquid media.
- Screen by restriction digest (optional)
- Amplify the target region.
- Digest the PCR product with the restriction enzyme that coincides with the sgRNA cut site/resides within the expected deletion site.
- Run fragments on an agarose gel. If CRISPR-Cas has induced a mutation, then the restriction site will no longer be functional and fragments will remain uncut. Take care to amplify fragments or use restriction enzymes that give a unique restriction site. Otherwise several small fragments, which are difficult to visualise may occur, regardless of mutation.
- If enriching for edited sequences, extract genomic DNA and perform the restriction digest. PCR amplify the target. Only WT sequences should be cut, allowing preferential amplification of the mutant fragment.
- Amplify the target region.
- Screen by sequencing
- Sequence PCR products to verify deletions (Figure 2) using a service such as the Eurofins Mix2seq kit.
- It can also be worth sequencing products which appear to have a WT sequence based on size. This can identify mutants which arise from activity of one sgRNA, leading to smaller indels at only one site. An example of this can be seen with M1 (Figure 2).
- Sequence PCR products to verify deletions (Figure 2) using a service such as the Eurofins Mix2seq kit.
- The protocol describes creation and assembly of all modules so that researchers can modify the overall system if desired, however, several of the L0 and L1 modules described are already available from Addgene at https://www.addgene.org/Thomas_Mock/. In addition, modules for P. tricornutum will soon be available from Addgene on the same website.
Data analysis
- Screening is carried out using the band shift assay and/or restriction site loss as described above. This allows assay products to be run on a gel to easily determine genome editing either by deletion (band shift assay) or mutation of the target site (restriction site loss). Products are then sequenced to confirm deletions/mutations.
- For primary clones around 30-40 colonies are tested for both Cas9 and deletions.
Note: We sequenced PCR products from all primary colonies, irrespective of the presence of Cas9 or deletions, and found that 11% of clones contained Cas9. 100% of clones with Cas9 contained a mutation–75% of the mutant clones contained deletions but the remaining 25% showed a mutation at one target site with a band similar to that of the WT. As a result, it can be worth screening for Cas9 as an indicator for potential mutations, as editing may not always result in a deletion. Cas9 clones can then be sub-cloned as described and subjected to further analysis including restriction site loss and sequencing. - Around 30 sub-clones are typically tested by band shift assay to determine the bi-allelic deletion frequency. This is calculated as the number of sub-clones with only the lower band (indicative of a deletion in both alleles), compared to the total number of sub-clones tested. Other colonies may give only the higher WT band or a mixture of both.
- Further analysis is dependent on which gene/sequence is being targeted. For example, when targeting the urease gene (which allows cells to use urea as a nitrogen source) in T. pseudonana, cell-lines were grown in media with either nitrate (positive control) or urea as the sole nitrogen source (Hopes et al., 2016). Cell counts and cell size were measured once a day until stationary phase was reached. Growth curves/cell size was then compared between cell lines (WT and mutant) and growth conditions (nitrate or urea) to determine nitrogen starvation or limitation in edited cell lines.
Recipes
- LB media/LB agar

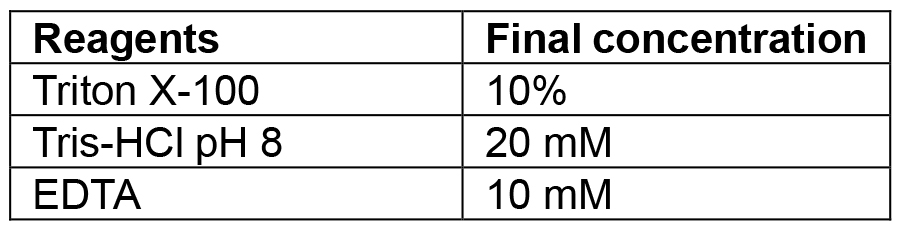
Combine tryptone, yeast extract and sodium chloride. If making plates also add agar. Make up to 1 L with MilliQ/distilled water and autoclave. If making selective media add the relevant antibiotic once the media has cooled to 50 °C. Plates are also poured once media has reached 50 °C. The recipe can be scaled up or down as needed - Lysis buffer
Acknowledgments
Thanks to The Sainsbury Laboratory for supplying the Golden Gate destination vectors and linkers. This work has been funded by a PhD studentship from the Natural Environment Research Council (NERC) awarded to Amanda Hopes. TM acknowledges partial funding from NERC (NE/K004530/1) and the School of Environmental Sciences at University of East Anglia, Norwich.
Author’s contributions: AH and TM conceived the project. AH designed and developed the protocol with input on Golden Gate cloning and the band shift assay method from Vladimir Nekrasov. AH and NB determined the U6 promoter for F. cylindrus and IG tested its activity in the same species. AH wrote the paper.
This protocol is based on the work presented in Hopes et al. (2016).
The authors declare that they have no competing interests.
References
- Belhaj, K., Chaparro-Garcia, A., Kamoun, S. and Nekrasov, V. (2013). Plant genome editing made easy: targeted mutagenesis in model and crop plants using the CRISPR/Cas system. Plant Methods 9(1): 39.
- Brooks, C., Nekrasov, V., Lippman, Z. B. and Van Eck, J. (2014). Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol 166(3): 1292-1297.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Engler, C., Youles, M., Gruetzner, R., Ehnert, T. M., Werner, S., Jones, J. D., Patron, N. J. and Marillonnet, S. (2014). A golden gate modular cloning toolbox for plants. ACS Synth Biol 3(11): 839-843.
- Hopes, A., Nekrasov, V., Kamoun, S. and Mock, T. (2016). Editing of the urease gene by CRISPR-Cas in the diatom Thalassiosira pseudonana. Plant Methods 12: 49.
- Kroth, P. G. (2007). Genetic transformation: a tool to study protein targeting in diatoms. Methods Mol Biol 390: 257-267.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Nymark, M., Sharma, A. K., Hafskjold, M. C., Sparstad, T., Bones, A. M. and Winge, P. (2017). CRISPR/Cas9 gene editing in the marine diatom Phaeodactylum tricornutum. Bio Protoc e2442.
- Nymark, M., Sharma, A. K., Sparstad, T., Bones, A. M. and Winge, P. (2016). A CRISPR/Cas9 system adapted for gene editing in marine algae. Sci Rep 6: 24951.
- Pinto, F. L. and Lindblad, P. (2010). A guide for in-house design of template-switch-based 5' rapid amplification of cDNA ends systems. Anal Biochem 397(2): 227-232.
- Poulsen, N., Chesley, P. M. and Kröger, N. (2006). Molecular genetic manipulation of the diatom Thalassiosira pseudonana (Bacillariophyceae). J Phycolo 42(5):1059-1065.
- Price, N. M., Harrison, G. I., Hering, J. G., Nirel, P. M., Palenik, B. and Morel, F. (1989). Preparation and chemistry of the artificial algal culture medium aquil. Biological Oceanography 6: 443-461.
- Rastogi, A., Murik, O., Bowler, C. and Tirichine, L. (2016). PhytoCRISP-Ex: a web-based and stand-alone application to find specific target sequences for CRISPR/CAS editing. BMC Bioinformatics 17(1): 261.
- Sakuma, T., Nishikawa, A., Kume, S., Chayama, K. and Yamamoto, T. (2014). Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci Rep 4: 5400.
- Weber, E., Engler, C., Gruetzner, R., Werner, S. and Marillonnet, S. (2011). A modular cloning system for standardized assembly of multigene constructs. PLoS One 6(2): e16765.
- Zheng, Q., Cai, X., Tan, M. H., Schaffert, S., Arnold, C. P., Gong, X., Chen, C. Z. and Huang, S. (2014). Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 57(3): 115-124.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hopes, A., Nekrasov, V., Belshaw, N., Grouneva, I., Kamoun, S. and Mock, T. (2017). Genome Editing in Diatoms Using CRISPR-Cas to Induce Precise Bi-allelic Deletions. Bio-protocol 7(23): e2625. DOI: 10.21769/BioProtoc.2625.
Category
Microbiology > Microbial genetics > Mutagenesis
Plant Science > Phycology > DNA > Genome editing
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.