- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Markerless Gene Editing in the Hyperthermophilic Archaeon Thermococcus kodakarensis
(*contributed equally to this work) Published: Vol 7, Iss 22, Nov 20, 2017 DOI: 10.21769/BioProtoc.2604 Views: 8628
Reviewed by: Modesto Redrejo-RodriguezTimo LehtiAlba Blesa
Original research article
The authors used this protocol in:

Apr 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The advent of single cell genomics and the continued use of metagenomic profiling in diverse environments has exponentially increased the known diversity of life. The recovered and assembled genomes predict physiology, consortium interactions and gene function, but experimental validation of metabolisms and molecular pathways requires more directed approaches. Gene function–and the correlation between phenotype and genotype is most obviously studied with genetics, and it is therefore critical to develop techniques permitting rapid and facile strain construction. Many new and candidate archaeal lineages have recently been discovered, but experimental, genetic access to archaeal genomes is currently limited to a few model organisms. The results obtained from manipulating the genomes of these genetically-accessible organisms have already had profound effects on our understanding of archaeal physiology and information processing systems, and these continued studies also help resolve phylogenetic reconstruction of the tree of life. The hyperthermophilic, planktonic, marine heterotrophic archaeon Thermococcus kodakarensis, has emerged as an ideal genetic system with a suite of techniques available to add or delete encoded activities, or modify expression of genes in vivo. We outline here techniques to rapidly and markerlessly delete a single, or repetitively delete several, continuous sequences from the T. kodakarensis genome. Our procedure includes details on the construction of the plasmid DNA necessary for transformation that directs, via homologous recombination, integration into the genome, identification of strains that have incorporated plasmid sequences (termed intermediate strains), and confirmation of plasmid excision, leading to deletion of the target gene in final strains. Near identical procedures can be employed to modify, rather than delete, a genomic locus.
Keywords: Genome editingBackground
Archaea often thrive in seemingly inhospitable and rapidly changing environments. Analyses of archaeal genomes reveal a plethora of metabolic strategies, predict sophisticated and highly interdependent regulatory networks underlying gene expression and reveal many genes whose protein–and increasingly often stable RNA–products lack a defined function. The ability to challenge existing, and define new pathways through genetic manipulation has assisted in deconvoluting archaeal physiology and information processing systems, and has more recently opened archaeal species to synthetic- and systems-level approaches to define intra- and intercellular networks.
Thermococcus kodakarensis is a hyperthermophilic, anaerobic, marine archaeon for which a genetic system has been developed over the last decade (Sato et al., 2003 and 2005; Fukui et al., 2005; Santangelo et al., 2008; Santangelo and Reeve, 2011; Hileman and Santangelo, 2012). The ability to genetically modify T. kodakarensis has allowed for the study of individual gene function in metabolism, replication, transcription and translation. Using a recombination based system and both selective and counter-selective markers, individual genes are deleted from the T. kodakarensis genome in a markerless manner (Figure 1). This markerless deletion strategy allows the consecutive deletion of multiple genes in a single strain using the same strategy for each gene.
T. kodakarensis strain TS559 (ΔTK2276; ΔTK0254::TK2276; ΔTK0149; ΔTK0664) requires the presence of agmatine and tryptophan for cellular growth (Santangelo et al., 2010). The deletion strategy presented here utilizes the selectable and counter-selectable markers TK0149 and TK0664, respectively. TK0149 encodes a pyruvoyl-dependent arginine decarboxylase, an enzyme necessary in the conversion of arginine to agmatine which is then converted to putrescine. Cells lacking TK0149 are dependent on the addition of agmatine to the media for viability. TK0664 encodes a hypoxanthine guanine phosphoribosyltransferase, an enzyme involved in a ribonucleotide scavenging pathway. Cells encoding TK0664 can metabolize 6-methylpurine (6-MP), a cytotoxic purine derivative, and thus perish in environments containing 6-MP. To assist others in implementing this technology, here we outline a procedure to delete a gene [as one example, we delete TK0566 (Walker et al., 2017)] from the T. kodakarensis TS559 genome. 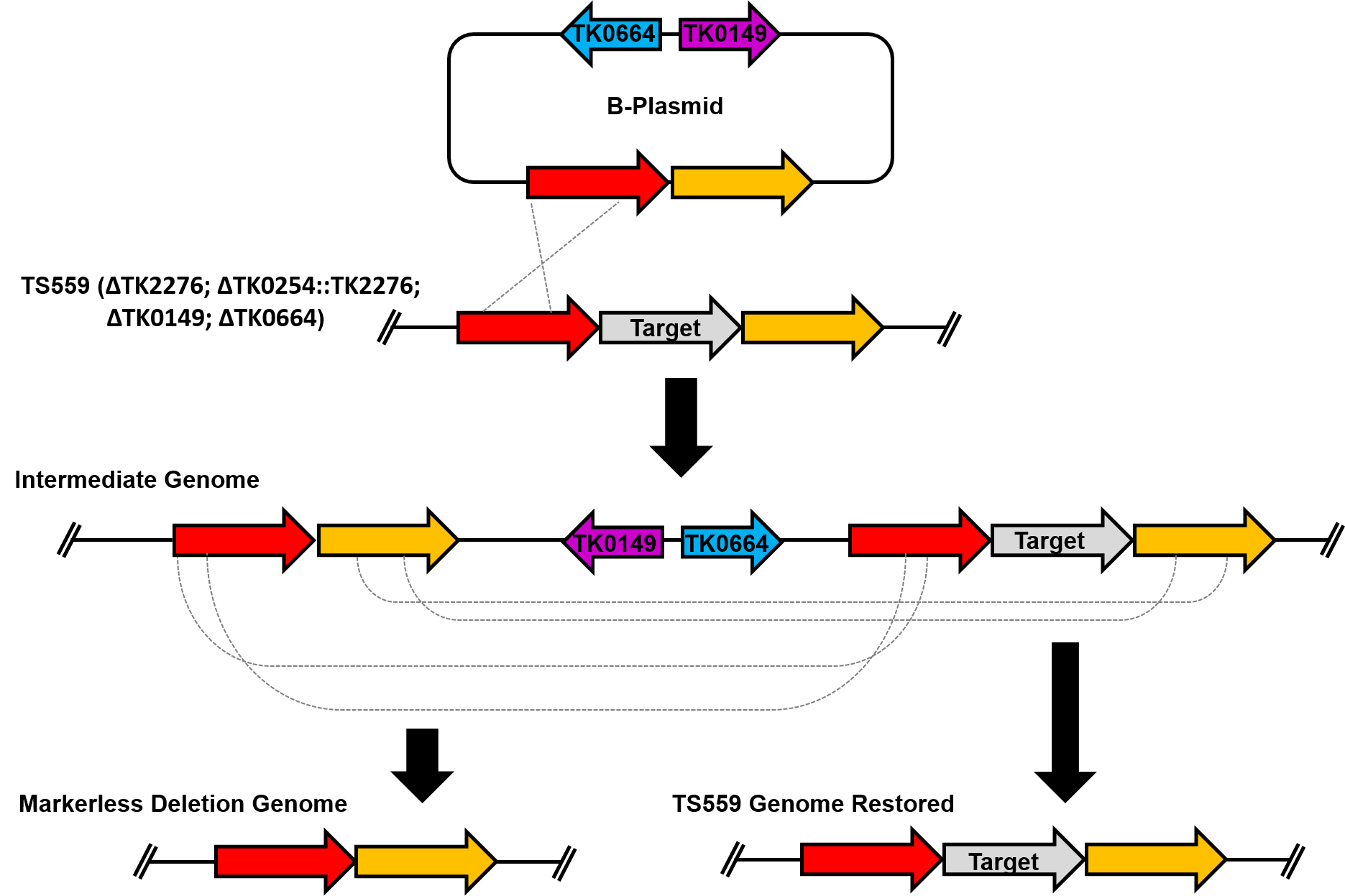
Figure 1. Overview of the markerless deletion scheme used in T. kodakarensis. At the top of the figure is the B-plasmid used to delete the target gene from the genome. The plasmid recombines into the genome providing agmatine prototrophy to recipient cells and yields an intermediate genome. Two intermediate genomes are possible; however only one is depicted here. A second spontaneous recombination event excises plasmid sequences and permits survival in the presence of cytotoxic 6-MP. This second recombination event will result in the desired deletion genome (left) or the restoration of the TS559 genome (right).
Materials and Reagents
- 1 ml TB syringe (BD, catalog number: 309624 )
- 1.7 ml microcentrifuge tubes (VWR, catalog number: 490004-444 )
- 0.2 ml PCR tubes (VWR, catalog number: 20170-012 )
- Polystyrene Petri plates (Fisher Scientific, catalog number: S33580A )
- Split rubber stopper (DWK Life Sciences, Wheaton, catalog number: W224100-282 )
- 20 mm aluminum seals (DWK Life Sciences, Wheaton, catalog number: 224178-01 )
- 20 mm E-Z Crimper, Standard Seal (DWK Life Sciences, Wheaton, catalog number: W225303 )
- 20 mm E-Z Decapper (DWK Life Sciences, Wheaton, catalog number: W225353 )
- Polycarbonate centrifuge tubes (Beckman Coulter, catalog number: 361690 )
- Cell spreader (Fisher Scientific, catalog number: 08-100-10 )
- 10 ml serum bottles (DWK Life Sciences, Wheaton, catalog number: 223739 )
- Face shields, lab coats, and autoclave gloves
- Paper towels
- T. kodakarensis strain TS559 (Santangelo et al., 2010)
- DH5α E. coli competent cells (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18258012 )
- XL1-Blue E. coli competent cells (Agilent Technologies, catalog number: 200228 )
- pTS700 (Hileman and Santangelo, 2012)
Note: Please contact corresponding author to obtain plasmid. - Agmatine (Sigma-Aldrich, catalog number: A7127 )
- Elemental sulfur (Aqua Solutions, catalog number: S8800-12KG )
- 10 mM Tris-HCl pH 8.0 (VWR, catalog number: 97061-258 )
- Isopropanol (Sigma-Aldrich, catalog number: 190764 )
- LE Quick dissolve agarose (VWR, catalog number: 490000-004 )
- Ethidium bromide (Sigma-Aldrich, catalog number: E1510 )
- AMPure XP (Fisher Scientific, catalog number: NC9959336)
Manufacturer: Beckman Coulter, catalog number: A63880 . - Nucleospin Gel and PCR Clean-up Kit (MACHERY-NAGEL, catalog number: 740609 )
- ZR Plasmid Miniprep kit (Zymo Research, catalog number: D4015 )
- T4 DNA polymerase (New England Biolabs, catalog number: M0203 )
- dGTP (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10297018 )
- SwaI restriction enzyme (New England Biolabs, catalog number: R0604 )
- dCTP (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10297018 )
- NEBuffer 2.1 (New England Biolabs, catalog number: B7202S )
- Taq DNA polymerase (New England Biolabs, catalog number: M0267 )
- dNTPs (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10297018 )
- 700Forward primer (5’ CGCCGCAATAGCGGTCGTCGTCATGTTCCC 3’)
- 700Reverse primer (5’ AACAATTTCACACAGGAAACAGCTATGACC 3’)
- Plasmid Miniprep kit
- Quikchange II (Agilent Technologies, catalog number: 200523 )
- DpnI
- Gelzan (Sigma-Aldrich, catalog number: G1910 )
- Phusion DNA polymerase (New England Biolabs, catalog number: M0530 )
- 6-Methylpurine (Sigma-Aldrich, catalog number: M1256 )
Note: This product has been discontinued. - Polysulfides
- Phenol (AMRESCO, catalog number: 0945 )
- Chloroform (Sigma-Aldrich, catalog number: C2432 )
- Isoamyl alcohol (EMD Millipore, catalog number: 1009791000 )
- Tryptone (EMD Millipore, catalog number: 1072131000 )
Note: T. kodakarensis requires casein peptone that is enzymatically digested using pancreatic enzymes. Other sources of tryptone are suitable for E. coli media. - Yeast extract (AMRESCO, catalog number: J850 )
Note: For E. coli media, any yeast extract is suitable, however T. kodakarensis requires this source of yeast extract. - Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 793566 )
- Agar
- Ampicillin (Sigma-Aldrich, catalog number: A0166 )
- Niacin (Sigma-Aldrich, catalog number: PHR1276 )
- Biotin (AMRESCO, catalog number: 0340 )
- Pantothenate (Sigma-Aldrich, catalog number: 259721 )
Note: This product has been discontinued. - Lipoic acid (Fisher Scientific, catalog number: BP26821 )
Note: This product has been discontinued. - Folic acid (Sigma-Aldrich, catalog number: F7876 )
- P-aminobenzoic acid (Acros Organics, catalog number: 146210010 )
- Thiamine (Fisher Scientific, catalog number: BP892-100 )
- Riboflavin (Sigma-Aldrich, catalog number: R1706 )
Note: This product has been discontinued. - Pyridoxine (Sigma-Aldrich, catalog number: P9755 )
- Cobalamin (Sigma-Aldrich, catalog number: V6629 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M9272 )
- Magnesium sulfate heptahydrate (MgSO4·7H2O) (EMD Millipore, catalog number: MX0070 )
- Ammonium sulfate ((NH4)2SO4) (VWR, catalog number: BDH9216 )
- Sodium bicarbonate (NaHCO3) (Sigma-Aldrich, catalog number: S6014 )
- Calcium chloride dihydrate (CaCl2·2H2O) (EMD Millipore, catalog number: CX0130 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P3911 )
- Potassium phosphate monobasic (K2HPO4) (Sigma-Aldrich, catalog number: P0662 )
- Sodium bromide (NaBr) (Fisher Scientific, catalog number: S255 )
- Strontium chloride hexahydrate (SrCl2·6H2O) (Sigma-Aldrich, catalog number: 13909 )
Note: This product has been discontinued. - Ammonium iron(II) sulfate hexahydrate (Fe(NH4)2(SO4)2·6H2O) (Sigma-Aldrich, catalog number: F3754 )
- Manganese(II) sulfate monohydrate (MnSO4·H2O) (Fisher Scientific, catalog number: M114 )
- Cobalt(II) chloride hexahydrate (CoCl2·6H2O) (Sigma-Aldrich, catalog number: 202185 )
- Zinc sulfate heptahydrate (ZnSO4·7H2O) (RICCA Chemical, catalog number: RDCZ0200 )
- Copper(II) sulfate pentahydrate (CuSO4·5H2O) (Sigma-Aldrich, catalog number: C8027 )
- Aluminum potassium sulfate dodecahydrate (AlK(SO4)2·12H2O) (Fisher Scientific, catalog number: A605 )
- Boric acid (H3BO3) (Fisher Scientific, catalog number: A73 )
- Sodium molybdate dehydrate (Na2MoO4·2H2O) (Sigma-Aldrich, catalog number: S6646 )
Note: This product has been discontinued. - Sodium sulfide nonahydrate (Na2S·9H2O) (Sigma-Aldrich, catalog number: 431648 )
- Cysteine (Fisher Scientific, catalog number: BP377 )
- Glutamic acid (AMRESCO, catalog number: 0421 )
- Glycine (Sigma-Aldrich, catalog number: W328707 )
- Arginine (Sigma-Aldrich, catalog number: A5131 )
- Proline (Sigma-Aldrich, catalog number: W331902 )
- Asparagine (AMRESCO, catalog number: 94341 )
- Histidine (Sigma-Aldrich, catalog number: H8000 )
- Isoleucine (Sigma-Aldrich, catalog number: W527602 )
- Leucine (Sigma-Aldrich, catalog number: L8000 )
- Lysine (Sigma-Aldrich, catalog number: L5626 )
- Threonine (Sigma-Aldrich, catalog number: T8625 )
- Tyrosine (Sigma-Aldrich, catalog number: T3754 )
- Alanine (Sigma-Aldrich, catalog number: W381829 )
- Methionine (Sigma-Aldrich, catalog number: M9625 )
- Phenylalanine (Sigma-Aldrich, catalog number: P2126 )
- Serine (Sigma-Aldrich, catalog number: S8407 )
Note: This product has been discontinued - Tryptophan (AMRESCO, catalog number: E800 )
- Aspartic acid (AMRESCO, catalog number: 0192 )
- Glutamine (Sigma-Aldrich, catalog number: G3126 )
- Valine (Sigma-Aldrich, catalog number: V0500 )
- Phenol:Chloroform:Isoamyl Alcohol (25:24:1) (see Recipes)
- LB plates (see Recipes)
- LB-Amp plates (see Recipes)
- KOD Vitamins (see Recipes)
- ASW-YT media (see Recipes)
- 2x ASW solution (see Recipes)
- Trace minerals solution (see Recipes)
- Polysulfides (see Recipes)
- 0.8x ASW solution (see Recipes)
- 20 amino acid solution (see Recipes)
Equipment
- Eppendorf Microcentrifuge 5424 (Eppendorf, model: 5424 )
- 125 ml serum bottles (DWK Life Sciences, Wheaton, catalog number: 223748 )
- Noodle pot
- Autoclave
- Anaerobic chamber (Coy Labs)
- Glass Petri plates (VWR, catalog number: 89000-304 )
Note: Glass Petri plates are used here, as many plastic Petri plates melt at T. kodakarensis incubation temperature (85 °C). - Beckman Avanti J Series centrifuge system
- JLA10.500 rotor (Beckman Coulter, model: JLA10.500 , catalog number: 369681)
- Eppendorf Mastercycler Nexus Thermal cycler (Eppendorf, catalog number: 6333000022 )
- GasPak EZ Anaerobe Container System (BD, model: GasPakTM EZ, catalog number: 260678)
- Pipettes (Gilson, catalog numbers: F123600 , F123615 , F123602 )
- Enzyme Cooler, Isotherm System (Eppendorf, model: IsoTherm-System® start set, catalog number: 3880000011 )
- VWR forced air incubator (37 °C and 85 °C)
- Thermo MaxQ 4000 benchtop orbital shaker (Thermo Fisher Scientific, Thermo ScientificTM, model: MaxQTM 4000 )
- Dry block heater (VWR, catalog number: 12621-090 )
Software
- Primer3 (Untergasser et al., 2007)
Procedure
- Primer design
- The first step in constructing a deletion strain is generating a plasmid construct that will facilitate the deletion of the gene of interest, referred to here as your favorite gene (YFG). A specific amplicon must be inserted into a common plasmid backbone (pTS700, see below), and the amplicon of choice must be generated with specific 5’ and 3’ sequences (added to the primers that generate the amplicon) to facilitate construction of the desired plasmid. Primer design starts by identifying the sequence of your favorite gene (YFG) as well as the sequence of adjacent regions in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa et al., 2017). The T. kodakarensis reference genome is entry T00226.
- Copy the positive-strand sequences encoding YFG plus 700 nucleotides upstream and 700 nucleotides downstream of YFG. End-join the 700 nucleotide sequence upstream and the 700 nucleotide sequence downstream of YFG by replacing the sequence of YFG with a single ‘N’. The resulting 1,401 nucleotide sequence serves as an input sequence for the primer design software program Primer3 (Untergasser et al., 2007).
- Within Primer3, set the parameters to demand selection of a pair of 25 nucleotide primers that will yield an amplicon of ≥ 1,200 nucleotides from the inputted 1,401 nucleotide sequence. Each primer should have a minimum Tm of 57 °C, no maximum Tm, 40%-75% G/C, and a 2 nucleotide GC-3’ end clamp. Primer3 typically returns multiple primer pairs, and for convenience only, we select the first pair. These algorithm derived primers are sufficient to amplify the amplicon of choice, but must be amended to add sequences that facilitate insertion into pTS700.
- To the upstream primer, add the sequence 5’ GGAGGTGAATTTC. This new primer (38 nucleotides) will be designated 001-XXXX, where XXXX is the gene number. To the downstream primer, 5’ GGTGAAGGATTTC will be added, and this primer will be designated 002-XXXX.
Note: The additional sequence added to 001-XXXX and 002-XXXX primers facilitates the use of Ligation Independent Cloning (LIC), our preferred cloning method, and may be omitted if a different cloning method is used.
- The first step in constructing a deletion strain is generating a plasmid construct that will facilitate the deletion of the gene of interest, referred to here as your favorite gene (YFG). A specific amplicon must be inserted into a common plasmid backbone (pTS700, see below), and the amplicon of choice must be generated with specific 5’ and 3’ sequences (added to the primers that generate the amplicon) to facilitate construction of the desired plasmid. Primer design starts by identifying the sequence of your favorite gene (YFG) as well as the sequence of adjacent regions in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa et al., 2017). The T. kodakarensis reference genome is entry T00226.
- A-plasmid construction
Note: Construction of an A-plasmid is not necessary to delete a gene from the T. kodakarensis genome. The A-plasmid serves as a building block from which multiple gene modification constructs can be built, including the addition of an affinity tag sequence, truncated versions of the gene, or allelic changes within the gene. Thus, if the deletion of the gene is the only desired result, B-plasmid construction (see ‘Mutagenic PCR for B-plasmid construction’) is the only necessary plasmid and may be constructed using an alternative cloning strategy. For example, ordering of a synthetic DNA fragment containing only the upstream and downstream regions of homology to YFG for use in cloning will suffice.- T. kodakarensis strain TS559 genomic DNA is prepared for amplification of the gene of interest using the primers designed above (001-XXXX and 002-XXXX). To properly inoculate a fresh culture of T. kodakarensis, 1 ml culture from a stock, 100 μl 1 M agmatine, 100 μl KOD Vitamins (see Recipes), and 0.2 g of sulfur are added to 100 ml of ASW-YT media (see Recipes) under anaerobic conditions. The newly inoculated culture is incubated at 85 °C for 12 h prior to the transformation. Using a 1 ml syringe, remove 1 ml of T. kodakarensis TS559 culture and transfer to a 1.7 ml microcentrifuge tube, and collect the cells via centrifugation (9,000 x g for 5 min in a tabletop centrifuge). Decant the supernatant, resuspend the harvested cells in 100 µl of 10 mM Tris-HCl pH 8.0, and then add 50 μl of Phenol:Chloroform:Isoamyl Alcohol (see Recipes). After vigorously mixing, separate organic and aqueous phases via centrifugation (9,000 x g, 5 min in the tabletop centrifuge), aliquot 50 μl of the upper aqueous layer to a fresh 1.7 ml microcentrifuge tube that contains 50 μl 10 mM Tris-HCl pH 8.0. Precipitate the nucleic acids by adding 100 μl of 100% isopropanol. Following a 30 min spin (9,000 x g, tabletop centrifuge), carefully remove the isopropanol and allow the near-colorless, small nucleic acid pellet to dry for 10 min. Resuspend the dried pellet in 30 μl 10 mM Tris-HCl pH 8.0. The DNA is not quantified prior to use in PCR reactions.
- PCR amplification is typically achieved using 001-XXXX and 002-XXXX primers in a mixture (50-100 µl) containing 3 μl of the genomic DNA (~300 ng), as prepared above. Any high-fidelity DNA polymerase can be used for the PCR amplification and should be used following the manufacturer’s recommendations.
- To determine if the amplicon of interest was generated, ~5 µl of the amplification reaction is loaded into a 1% agarose gel, resolved, stained with ethidium bromide (EtBr), and imaged. If the correct product was amplified with no alternative products, the resultant amplicon is purified using AMPure XP beads following the manufacturer’s directions. If a mixture of desired and alternative products were identified, the total reaction should be loaded into the gel, resolved, stained, imaged and the desired amplicon should be excised out of the gel and purified using a commercially available gel purification kit such as Nucleospin Gel and PCR Clean-up Kit by Machery-Nagel following the manufacturer’s instructions.
Note: A gel purification kit or PCR clean-up kit can be used as an alternative to AMPure XP beads. - Simultaneous to generating the desired amplicon for YFG, the plasmid pTS700 should be prepared for accepting the amplicon. pTS700 is the vector used for all gene deletion constructs and contains an E. coli origin of replication (oriC), AmpR, and T. kodakarensis selectable and counter-selectable markers, TK0149 and TK0664, respectively. pTS700 also features a SwaI restriction enzyme cut site that is used to insert, via LIC, the YFG amplicon. pTS700 can be maintained in any standard E. coli strain using ampicillin as the selectable marker, is easily recovered from cultures via commercially available miniprep kits (we typically use the ZR Plasmid Miniprep Kit and follow manufacturer’s instructions), and the concentration determined by fluorometric or spectroscopic techniques.
- At this point, both the YFG desired amplicon and pTS700 should be purified. LIC will be used to build the new plasmid, pCSUXXXXA, again where XXXX is the gene number of YFG. LIC uses 12 nucleotide complimentary overhangs between the plasmid and PCR product to drive incorporation of the amplicon sequences into the vector. The complementarity is generated by T4 DNA polymerase (DNAP)-mediated exonuclease activity on the vector and amplicon sequences (Figure 2).
Note: LIC is not essential in A-plasmid construction. We utilize LIC for plasmid construction because it is the most cost-effective option for constructing deletion plasmids for all 2,306 genes in the T. kodakarensis genome. LIC is not dependent on a DNA ligase or phosphatase, and the only enzyme required is T4 DNAP (Aslanidis and de Jong, 1990). Any standard cloning method may be used in place of LIC.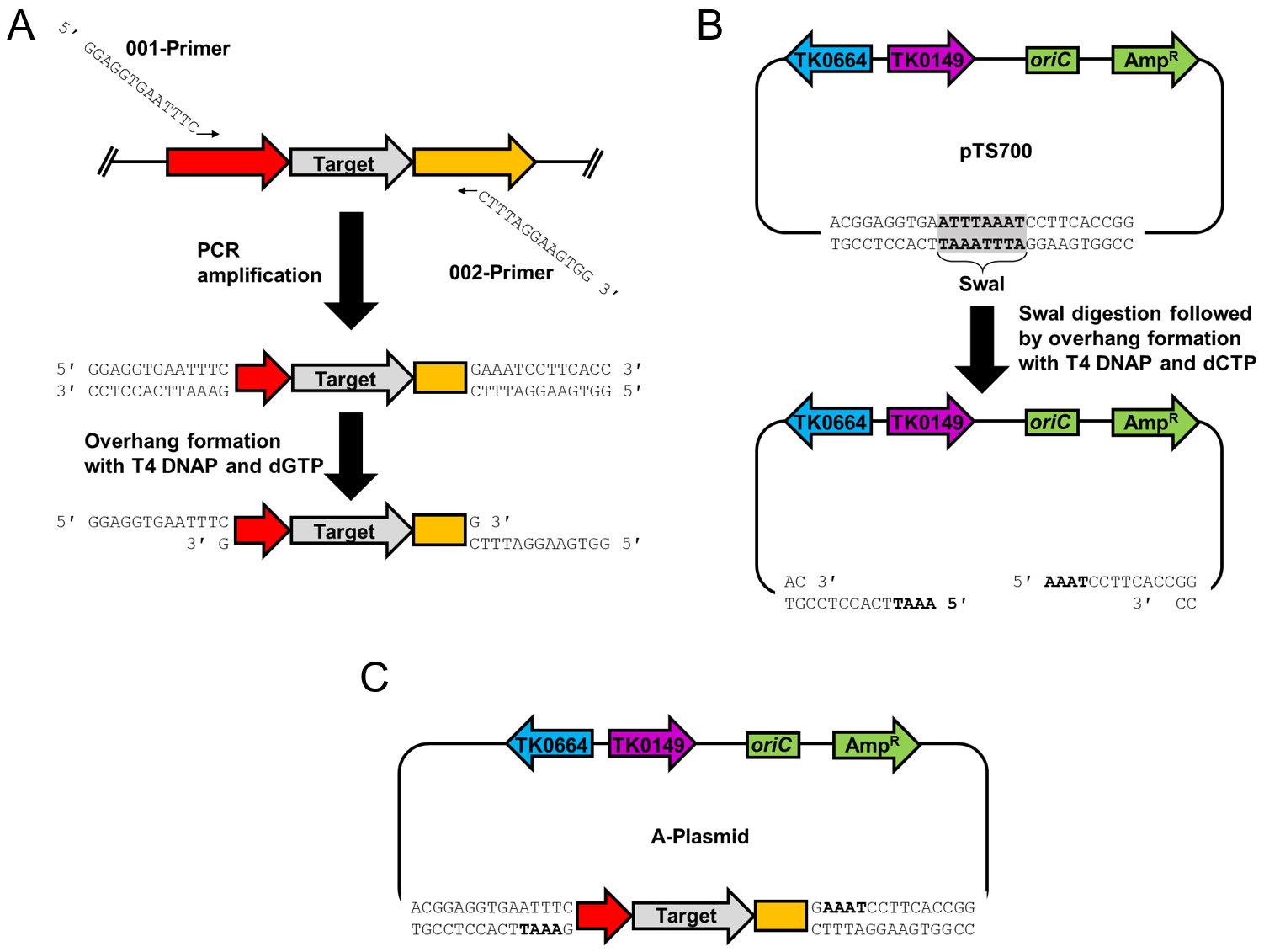
Figure 2. Construction of the A-plasmid using ligation independent cloning. A. The target gene as well as the upstream and downstream sequences are amplified using 001-XXXX and 002-XXXX primers that include the necessary 5’-tail sequences. The purified amplicon is incubated with T4 DNAP and dGTP to generate the 12 nucleotide overhangs. B. pTS700 is digested using SwaI and then incubated with T4 DNAP and dCTP to generate amplicon-complementary 12 nucleotide overhangs. C. The T4 DNAP treated amplicon and plasmid are incubated together and transformed into E. coli to generate the A-plasmid.
Digest 500 ng of purified pTS700 with SwaI in a 20 μl reaction following manufacturer’s instructions. Following SwaI digestion, 1 μl of T4 DNAP and 1 μl of 100 mM dCTP are added, incubation is continued at 37 °C for 30 min, then 20 min at 85 °C to heat inactivate all enzymes, and finally the reaction is placed on ice.
Simultaneously, a second reaction will prepare the YFG amplicon for LIC. Mix 500 ng desired amplicon, 0.9 μl NEBuffer 2.1, 0.5 μl 100 mM dGTP, and 0.5 µl T4 DNAP in a final volume of 9 μl, incubate at 37 °C for 30 min, then 20 min at 85 °C to heat inactivate the enzymes. 1 µl aliquots of each reaction are combined in a single tube, heated to 85 °C for 3 min, and the mixtures allowed to slowly cool to room temperature. This slow cooling permits the 12 nucleotide complementary overhangs to anneal. Following the cooling step, the 2 µl reaction is transformed into 50 μl competent cells of any standard E. coli strain, and spread onto LB-Amp plates (see Recipes). - After overnight incubation, colony PCR is used to determine if resultant transformants contain the newly constructed, desired A-plasmid. At least 10 distinct colonies should be checked using the pTS700 specific primers (700Forward and 700Reverse), although in some cases additional colonies may need to be screened. Each colony is picked from the plate into 6 μl of dH2O in a 0.2 ml PCR tube and resuspended. 4 μl is removed and spotted onto a LB-Amp plate leaving 2 μl behind in the PCR tube (ensure that the label on the PCR tube matches that on the spot plate). Once all colonies are spotted, incubate the LB + Amp plate at 37 °C to allow overnight growth. The remaining 2 μl of cells will be used as the DNA component in an amplification reaction using Taq DNAP. Taq DNAP yields the best results as it is not sensitive to the cellular and media components carried over into the reactions.
Colony PCR conditions (20 μl)Taq DNAP (20 U/μl) 0.1 μl dNTPs (2.5 mM) 1.6 μl 10x ThermoPol buffer 2 μl 700Forward primer (100 μM) 0.1 μl 700Reverse primer (100 μM) 0.1 μl Resuspended cells 2 μl H2O 13.9 μl
Following PCR, the total reaction is loaded into a 1% agarose gel, resolved, stained, and imaged. If the LIC was unsuccessful, a product of 150 bp will be generated. If LIC was successful, a product of 150 bp plus the size of your initial amplicon (YFG + 1,400 bp flanking regions) will be present. Identify one of the successful LIC generated plasmids, return to the spot plate, and pick the corresponding colony into 5 ml of LB with ampicillin and grow overnight at 37 °C while shaking. Purify the plasmid from E. coli using a preferred plasmid Miniprep kit, quantify, and sequence the entire amplicon using the 700Forward and 700Reverse primers in two separate reactions. If the sequencing confirms the YFG along with the upstream and downstream sequences have been inserted into pTS700, the plasmid is now designated pCSUXXXXA and is referred to as the A-plasmid.
- T. kodakarensis strain TS559 genomic DNA is prepared for amplification of the gene of interest using the primers designed above (001-XXXX and 002-XXXX). To properly inoculate a fresh culture of T. kodakarensis, 1 ml culture from a stock, 100 μl 1 M agmatine, 100 μl KOD Vitamins (see Recipes), and 0.2 g of sulfur are added to 100 ml of ASW-YT media (see Recipes) under anaerobic conditions. The newly inoculated culture is incubated at 85 °C for 12 h prior to the transformation. Using a 1 ml syringe, remove 1 ml of T. kodakarensis TS559 culture and transfer to a 1.7 ml microcentrifuge tube, and collect the cells via centrifugation (9,000 x g for 5 min in a tabletop centrifuge). Decant the supernatant, resuspend the harvested cells in 100 µl of 10 mM Tris-HCl pH 8.0, and then add 50 μl of Phenol:Chloroform:Isoamyl Alcohol (see Recipes). After vigorously mixing, separate organic and aqueous phases via centrifugation (9,000 x g, 5 min in the tabletop centrifuge), aliquot 50 μl of the upper aqueous layer to a fresh 1.7 ml microcentrifuge tube that contains 50 μl 10 mM Tris-HCl pH 8.0. Precipitate the nucleic acids by adding 100 μl of 100% isopropanol. Following a 30 min spin (9,000 x g, tabletop centrifuge), carefully remove the isopropanol and allow the near-colorless, small nucleic acid pellet to dry for 10 min. Resuspend the dried pellet in 30 μl 10 mM Tris-HCl pH 8.0. The DNA is not quantified prior to use in PCR reactions.
- Mutagenic PCR for B-plasmid construction
- The B-plasmid is used for gene deletions and lacks the YFG but retains the upstream and downstream sequences. To construct the B-plasmid, a Quikchange-II reaction is used with the A-plasmid as DNA template. 60 nucleotide primers are employed, and the primers bind to 30 nucleotides on either side of the gene of interest thus deleting the sequences encoding YFG from the final product. The first 30 nucleotides should be identical to the 30 nucleotides immediately upstream of YFG, while the 3’ terminal 30 nucleotides should be identical to the 30 nucleotides immediately downstream of YFG. These long primers ensure the primers are of the adequate melting temperature (> 78 °C). The 60 nucleotide primers used in the Quikchange are designated 016-XXXX and 017-XXXX and, as Quikchange demands, the primers are reverse complements of each other.
- The conditions for the Quikchange reaction follow exactly the Quikchange-II kit protocol including the DpnI treatment to degrade the E. coli methylated A-plasmid. After the DpnI treatment, 2 μl of the total reaction is used to transform 50 μl XL1-Blue E. coli cells, which are then plated on LB-Amp agar plates, and incubated overnight at 37 °C. Colony PCR is performed, again using primers 700Forward and 700Reverse, to determine if the sequences encoding YFG were deleted from the A-plasmid leaving only the now-fused upstream and downstream sequences. The product size for all deletions should be ~1,400 bp, and the colony corresponding to the deletion is picked into 5 ml LB-Amp, incubated at 37 °C overnight, and newly constructed B-plasmid DNA purified using the preferred plasmid Miniprep Kit. The resulting purified B-plasmid is sequenced using the 700Forward and 700Reverse primers in two separate reactions. If the sequencing demonstrates that YFG has been deleted, this new plasmid is now designated pCSU-XXXXB and will be used to construct the T. kodakarensis deletion strain.
- The B-plasmid is used for gene deletions and lacks the YFG but retains the upstream and downstream sequences. To construct the B-plasmid, a Quikchange-II reaction is used with the A-plasmid as DNA template. 60 nucleotide primers are employed, and the primers bind to 30 nucleotides on either side of the gene of interest thus deleting the sequences encoding YFG from the final product. The first 30 nucleotides should be identical to the 30 nucleotides immediately upstream of YFG, while the 3’ terminal 30 nucleotides should be identical to the 30 nucleotides immediately downstream of YFG. These long primers ensure the primers are of the adequate melting temperature (> 78 °C). The 60 nucleotide primers used in the Quikchange are designated 016-XXXX and 017-XXXX and, as Quikchange demands, the primers are reverse complements of each other.
- Thermococcus kodakarensis transformation
- The B-plasmid must be transformed into T. kodakarensis strain TS559. The first step of the transformation is to inoculate a fresh culture of TS559 as described in step B1.
- To complete the transformation, the cells will be spread onto specialized plates that will remain solid at 85 °C. The media used to make the plates must be autoclaved immediately before pouring. Plates should be poured no more than 24 h prior to use. Two 100 ml serum bottles with unique ingredients are autoclaved separately and mixed immediately before pouring. Each set of serum bottles will yield four plates, and each transformation requires two plates. For all plates, one of the bottles will remain consistent and contains 1 g of Gelzan in 50 ml of distilled H2O. Once the Gelzan and dH2O are added, cap the serum bottle with a rubber stopper and aluminum seal and shake to mix well. Note that the Gelzan will not completely dissolve prior to autoclaving. The contents of the second bottle will vary depending on the composition of the desired solid media. The media required for the initial transformation is ASW-YT-S, and thus the second serum bottle will contain 50 ml of 2x ASW (see Recipes) with 0.5 g yeast extract, 0.5 g tryptone, and 500 μl trace minerals (see Recipes). After all reagents are in the bottle, cap with a rubber stopper and seal with an aluminum seal.
Note: Place both bottles in an autoclave-safe vessel with a lid and add a small amount of water to the bottom. For this, we used a large noodle pot, however any large vessel with a lid will work. The sealed bottles present an explosion hazard in the autoclave. Placing the bottles in a large noodle pot ensures that on the rare occasions an explosion occurs it will be contained. When working with sealed vials and the autoclave, proper safety precautions should be taken including the use of face shields, lab coats, and autoclave gloves.
A standard liquid autoclave cycle with a 20-min sterilization at 121 °C is sufficient to sterilize the media and dissolve the Gelzan. As soon as the autoclave cycle has finished, remove the noodle pot, and bring the bottles into the anaerobic chamber. The Gelzan will begin to solidify at any temperature below 85 °C and will remain solidified so you must pour the plates immediately after the autoclave cycle has completed. For every set of two bottles, four, previously autoclaved, glass Petri plates should be arranged in the chamber to allow for quick pouring. Once the bottles are in the chamber, work quickly to uncap the bottle containing the 2x ASW and add to this bottle 100 μl Vitamins and 200 μl polysulfides (see Recipes). Swirl to mix well, careful not to spill. Now, uncap the bottle containing Gelzan and pour the 2x ASW bottle into the Gelzan bottle, again swirl to mix well. Pour approximately ¼ of the bottle into each plate. The plates should set in about 10 sec. Allow the plates to cool for ~10 min then flip the plates over until ready for use. - After 12 h, the TS559 liquid culture (100 ml) is taken into the anaerobic chamber and poured into a polycarbonate centrifuge tube suited for use in a high-speed centrifuge. The centrifuge tube is sealed, removed from the chamber, and spun in a JLA10.500 rotor at 18,000 x g for 10 min at 4 °C to pellet the cells. After centrifugation, immediately remove and invert the centrifuge bottle so that the cell pellet is not disturbed by the supernatant while moving the tube back into the anaerobic chamber. Once the tube is back in the chamber, carefully remove the supernatant without disturbing the pellet.
Note: Polycarbonate tubes must be used as they can withstand the high temperatures of the cultures. - The cell pellet is resuspended in 3 ml of 0.8x ASW (see Recipes). Each transformation only requires 200 μl of resuspended cells, therefore a 100 ml of culture yields enough cells for ~15 transformations. Following resuspension, remove the cells from the centrifuge bottle and place them in 1.7 ml microcentrifuge tubes on ice.
Notes:- To prevent the chambers from becoming humid, use a small ice bucket with a lid or an enzyme cooler. Allow the cells to incubate on ice for 30 min.
- Cells cannot be stored for future experiments.
- To prevent the chambers from becoming humid, use a small ice bucket with a lid or an enzyme cooler. Allow the cells to incubate on ice for 30 min.
- While the cells incubating on ice, the previously purified B-plasmid(s) are prepared for the transformation. For each transformation, aliquot 3 μg of purified B-plasmid into a 1.7 ml microcentrifuge tube and bring into the chamber.
- After the cells have incubated for 30 min on ice, add 200 μl of cells to the 3 μg of plasmid. Continue incubating on ice for an additional 60 min.
- Although T. kodakarensis is naturally competent, a heat shock step is used in the transformation protocol to increase the efficiency of the transformation. Heat the cell/plasmid mixture to 85 °C for 1 min followed by a 5 min recovery on ice. Each transformation will be spread on two plates, one with a high volume of cells (160 μl) and one with a low volume of cells (40 μl) using a cell spreader. After spreading, the plates are flipped upside down and placed in a metal cylinder, packed with paper towels to absorb moisture and a GasPak EZ Anaerobe Container System packet to maintain anaerobic conditions during growth. The cylinder is sealed in the chamber, removed from the chamber, and placed in an 85 °C incubator for 48-72 h.
- After allowing colony formation, remove the cylinder from the incubator and bring into the anaerobic chamber. Remove the plates from the cylinder and identify T. kodakarensis colonies. The colonies are small, clear, and can be difficult to identify. For this reason, it is difficult to pick single colonies directly from the transformation plates and it is therefore often necessary to spot the colonies. At least 10 colonies are spotted onto media of identical composition to the transformation plates. To spot single colonies, a colony from the transformation plate is picked into 6 µl of 0.8x ASW and serial dilutions are spotted (spots are ~6 µl). Using 0.8x ASW and a 10-fold dilution at each step, spot the colony for 10 dilutions onto freshly poured plates. The plates are then placed in a metal cylinder with an anaerobe pack and incubated at 85 °C for 48-72 h.
- Following the incubation, the plates are removed from the incubator and brought into the anaerobic chamber. One colony from each series of serial dilutions is used to inoculate 5 ml of ASW-YT supplemented with 0.1 g of elemental sulfur and 5 μl KOD Vitamins. These freshly inoculated bottles are sealed in the anaerobic chamber, removed, and incubated at 85 °C overnight. These cultures will be referred to as potential intermediate strains. After overnight growth, total genomic DNA will be extracted from these cultures as described in step B1.
- The B-plasmid must be transformed into T. kodakarensis strain TS559. The first step of the transformation is to inoculate a fresh culture of TS559 as described in step B1.
- Selection of intermediate strains
- For the transformation to be successful, indicated by the presence of viable colonies, the B-plasmid must have integrated into the genome via homologous recombination. The plasmid has the potential to recombine at one of two loci, either upstream or downstream of the gene of interest (Figure 3).
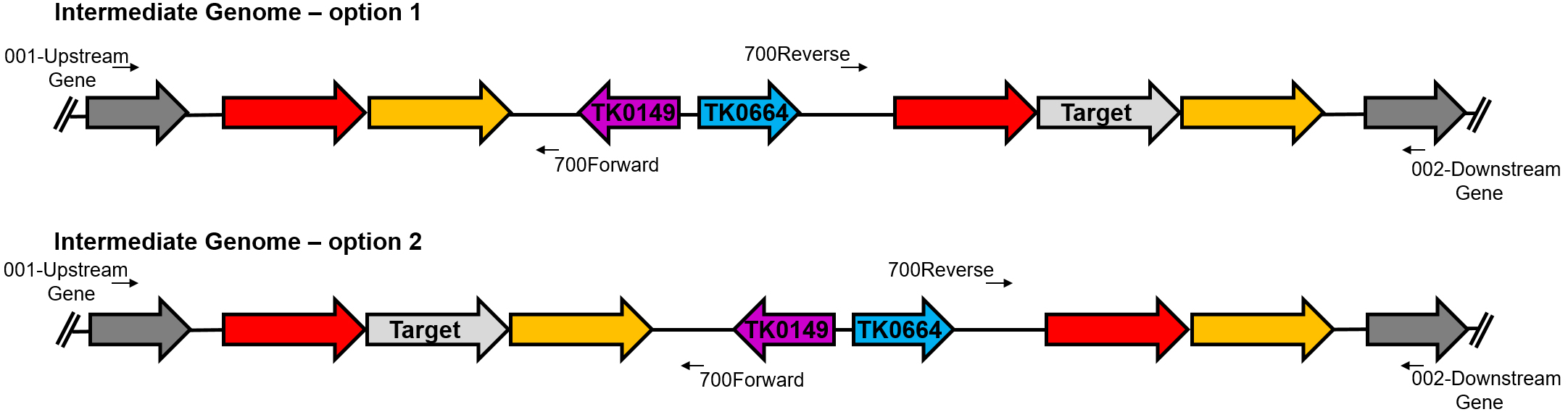
Figure 3. Identification of the intermediate genomes using PCR. The intermediate genomes are identified using primers specific to the B-plasmid (700Forward and 700Reverse) with primers specific to the genome sequence. For intermediate PCR, the 001 and 002 primers for the target gene cannot be used; instead the 001 and 002 primers for the upstream and downstream genes, respectively, should be used.
It is necessary to confirm via PCR that the B-plasmid has been integrated into the genome and determine the locus of this recombination. Purified genomic DNA from potential intermediate strains (as specified in step D9 above) will be used in PCR reactions with two separate primer combinations: 700Forward is used in combination with 002-XXXY while 700Reverse will be used 001-XXXW. PCR is performed using 3 μl (~300 ng) of genomic DNA with Phusion DNAP following manufacturer’s instructions. The PCR reactions are resolved in a 1% agarose gel, stained, and imaged. Ideally, both the upstream and downstream recombination events will be identified in individual colonies, however in some cases only one of the recombination events is identified.
Note: The 001 and 002 primers used in this PCR are not for YFG, but instead for the genes immediately upstream and downstream of YFG, respectively. If the 001 and 002 primers are not available for the adjacent genes, any primer should suffice as long as it is external to the 001 and 002 primers for the gene of interest. The 001 and 002 primers for the gene of interest cannot be used because they will bind both the plasmid and genomic DNA sequences. - Using the results from the intermediate PCR, two distinct intermediates are selected, ideally one each of the 2 possible recombination events. Each of the selected intermediate strains is used to inoculate 5 ml of ASW-YT supplemented with 5 μl KOD Vitamins, elemental sulfur, and 5 μl 1 M agmatine. Agmatine is added at this point so that the cells no longer have selective pressure to retain the plasmid in the genome and to allow for the second recombination event to occur. These cultures are allowed to grow for 12 h at 85 °C.
- To select for cells that have spontaneously excised the plasmid sequences and potentially generated the deletion strain of YFG, overnight cultures of the intermediate strains are spread on plates containing both agmatine and 6-methylpurine in addition to KOD Vitamins, polysulfides, and 20 amino acids solution (see Recipes). For each intermediate culture, two plates will be poured. As done in step D4, two 50 ml serum bottles are used. As before, one serum bottle will contain 1 g of Gelzan in 50 ml of H2O. The second serum bottle will now contain only 50 ml of 2x ASW and 500 μl trace minerals. The same autoclaving procedure is used. After removing the serum bottles from the autoclave and bringing each into the anaerobic chamber, the serum bottle containing 2x ASW is opened and 100 μl of 1 M agmatine, 100 μl 100 μM 6-methylpurine, 200 μl polysulfides, and 5 ml 20 amino acid solution are added and mixed well. This solution is then added to the Gelzan containing bottle, mixed well, and poured into 4 glass Petri plates.
- Following 12 h incubation, the agmatine-containing confirmed intermediate cultures are brought into the chamber where they will be spread onto the counter-selective plates. The two plates for each culture will be used to spread either high (160 μl) or low (40 μl) volumes of cells. For each plate, the proper number of cells are pipetted onto the surface and a cell spreader is used to spread the cells around the plate. Following this, the plates are put into a metal cylinder with an anaerobe pack and sealed. The cylinder is removed from the chamber and placed in the 85 °C incubator for 48-120 h.
- After 85 °C incubation, at least 10 colonies from the spread plates must be spotted onto newly poured plates made in the exact same manner as the spread plates to obtain single colonies. To spot single colonies, a colony from the spread the plate is picked into 6 μl of 0.8x ASW and serial dilution spot plating is performed using 0.8x ASW and a 10-fold dilution at each step for 10 steps (spots are ~6 µl). Following serial-dilution spotting, the plates are placed in a metal cylinder with an anaerobe pack and incubated at 85 °C for 48-120 h.
- After these spot plates have been allowed to grow, one colony from each serial dilution series is used to inoculate 5 ml of ASW-YT supplemented with 5 μl 1 M agmatine, 5 μl KOD Vitamins, and 0.1 g elemental sulfur. After overnight growth, total genomic DNA can be extracted from these cultures as described in step B1.
- For the transformation to be successful, indicated by the presence of viable colonies, the B-plasmid must have integrated into the genome via homologous recombination. The plasmid has the potential to recombine at one of two loci, either upstream or downstream of the gene of interest (Figure 3).
- Confirmation of deletion strain
- There are two possible recombination events that could occur when the plasmid recombines out of the genome. One recombination will return the genotype to the parental strain that retains YFG, while the other will yield the genome with the desired deletion of YFG. If after checking 30 individual colonies from at least 2 intermediates, ideally one from each of the possible recombination events, only the wildtype genome is observed, then the gene is deemed statistically essential. A series of different PCR reactions are performed to determine if YFG was deleted (Figure 4).
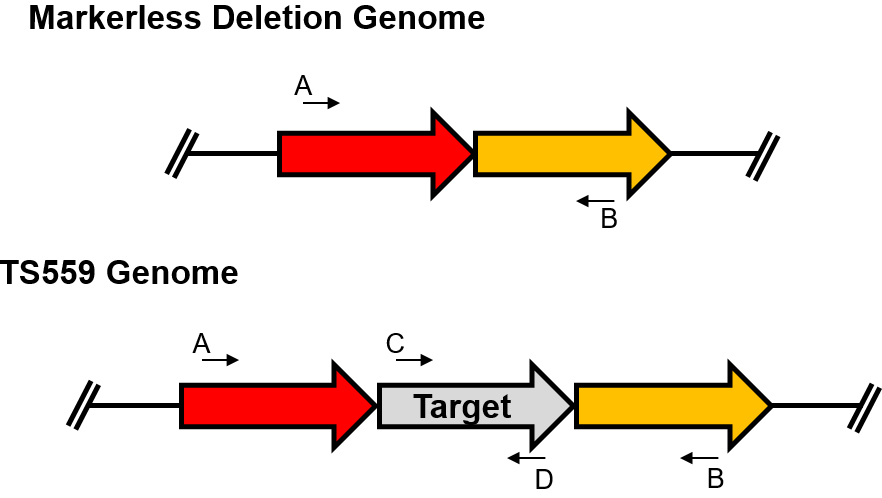
Figure 4. Confirmation of the desired deletion strain. The primer designated ‘A’ is the 001-XXXX and the primer designated ‘B’ is 002-XXXX. The C and D primers are any primers internal to the gene of interest. To confirm the deletion of the gene of interest, at least 3 combinations of primers must be used, A/B, C/D, and A/D or B/C. If the gene of interest has been deleted from the genome, no product will be synthesized using the C and D primers.
TS559 genomic DNA is always used as a positive control for the final PCR reactions. The first PCR uses the 001-XXXX and 002-XXXX primers to determine if YFG is deleted, and a second reaction uses either 001-XXXX or 002-XXXX and a primer internal to YFG. The third PCR reaction utilizes a pair of primers that are completely internal to YFG. The lack of a PCR product for the second and third reactions signifies the deletion occurred as long as the product was present using the control TS559 genomic DNA. These PCR reactions are performed using a high fidelity DNAP and 3 μl genomic DNA (~300 ng). Following PCR, the products are separated using a 1% agarose gel, stained with EtBr, and imaged. - If the desired deletion genome is identified within the final PCR amplifications, further amplifications are performed on that strain to extend confidence that YFG is truly deleted. The external PCR using the 001-XXXX and 002-XXX primers is repeated in a 50 μl reaction using Phusion DNAP, and 3 μl of purified genomic DNA (~300 ng). Following PCR, only 5 μl of this reaction is used in an agarose gel to confirm the PCR worked. The remaining reaction is sent to a DNA sequencing facility for purification and sequencing using the 001-XXXX and 002-XXXX primers in two separate sequence reactions.
- Once the sequencing has confirmed the deletion of YFG, the deletion strain has been constructed. If desired, a Southern blot or whole genome sequencing can be performed to further confirm the deletion.
- There are two possible recombination events that could occur when the plasmid recombines out of the genome. One recombination will return the genotype to the parental strain that retains YFG, while the other will yield the genome with the desired deletion of YFG. If after checking 30 individual colonies from at least 2 intermediates, ideally one from each of the possible recombination events, only the wildtype genome is observed, then the gene is deemed statistically essential. A series of different PCR reactions are performed to determine if YFG was deleted (Figure 4).
Data analysis
- Plasmid construction confirmation
Construction of both the A and B plasmid is typically a facile procedure with few problems. Complications can result from primer design errors or primer incompatibility, and when such occurs, the use of the second set of primers selected by the program is typically sufficient to resolve any issues. Infusion cloning can be used (following manufacturer’s instructions) in instances where LIC is unsuccessful. Construction of pCSU-0556A and pCSU-0556B were typical and non-problematic using the described protocol. - Intermediate strain PCR
PCR for the intermediate strains may only give a product for one of the primer pairs (001-XXXX/700Reverse or 002-XXXX/700Forward). In most cases, modification of the PCR conditions, annealing temperature or elongation time, will allow for amplification using both pairs of PCR primers. For the deletion of TK0556, both intermediate strains were identified using standard PCR conditions (Figure 5).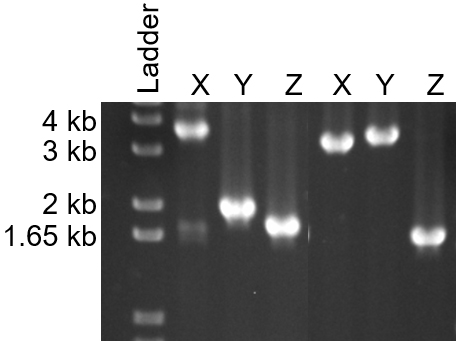
Figure 5. Identification of both intermediate genomes for the deletion of TK0566 using diagnostic PCR. X and Y lanes are used to determine the orientation of the intermediate genome. Primers 001-0565 and 700Reverse are used in the X reaction while 002-0567 and 700Forward are used in the Y reaction. If the genome is the first intermediate, the X product should be ~3,600 bp while the Y products should be ~1,600 bp. For the second intermediate, the X product should be ~3,600 bp and the Y product ~3,650 bp. A third, control PCR reaction (Z) was performed using 001-1418 and 002-1418 to ensure that the genomic preps yielded DNA suitable for PCR. The expected Z product was ~1,600 bp. - Final deletion strain PCR
Ideally, four PCR reactions, in addition to DNA sequencing, will be used to determine if the gene of interest was deleted from the T. kodakarensis TS559 genome (Figure 6). The PCR reaction using two primers internal to the gene of interest is crucial in ensuring that the gene is deleted from the genome, and has not moved to a different locus via an off-target recombination event.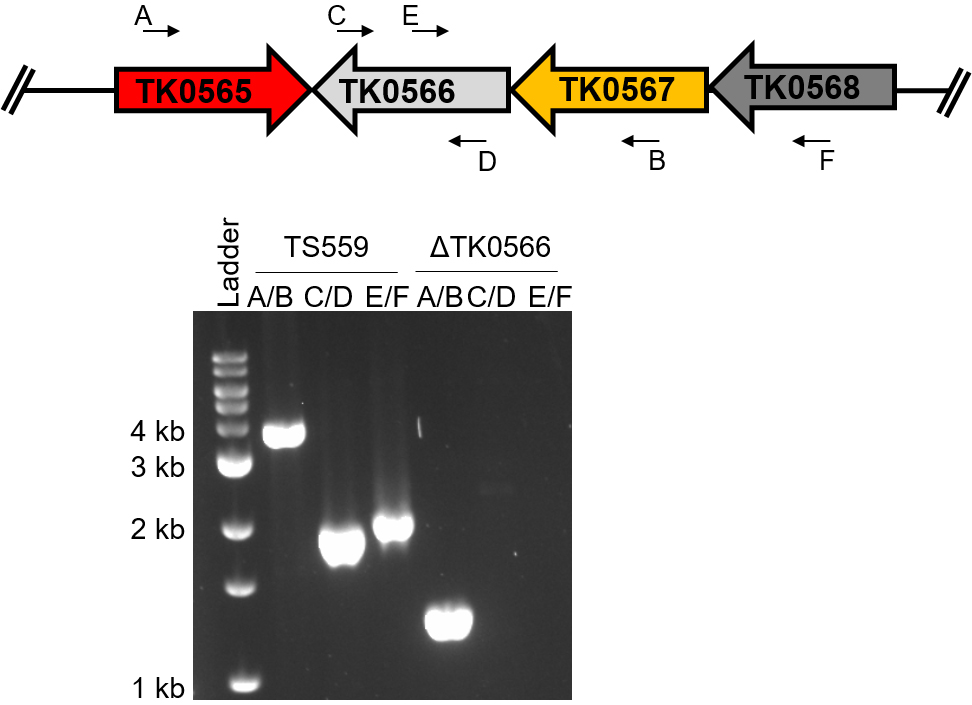
Figure 6. Diagnostic PCR confirms the deletion of TK0566 from the T. kodakarensis genome. The same primer pairs are used on the TS559 genome as the deletion genome. The size shift using primers A/B corresponds to the deletion of TK0566 while the absence of products for the reactions using C/D and E/F confirms the gene has been deleted from its native locus. The absence of products using primers C/D demonstrates that TK0566 is not present anywhere in the genome of the deletion strain.
Recipes
- Phenol:Chloroform:Isoamyl Alcohol (25:24:1)
25 ml Tris-saturated phenol
24 ml chloroform
1 ml isoamyl alcohol - LB media (1 L)
10 g tryptone
5 g yeast extract
10 g NaCl
Note: Must be autoclaved. - LB-Amp plates (1 L)
10 g tryptone
5 g yeast extract
10 g NaCl
15 g agar
Note: Must be autoclaved. - KOD Vitamins (200x) (1 L)
0.2 g niacin
0.08 g biotin
0.2 g pantothenate
0.2 g lipoic acid
0.08 g folic acid
0.2 g P-aminobenzoic acid
0.2 g thiamine
0.2 g riboflavin
0.2 g pyridoxine
0.2 g cobalamin
Note: This solution is light sensitive and should be protected. - ASW-YT media
Artificial sea-water medium supplemented with:
0.5% (w/v) tryptone
0.5% (w/v) yeast extract
1x trace mineral solution
1x Vitamin mixture
1x artificial sea-water contains (1 L):
20 g NaCl
3 g MgCl2·6H2O
6 g MgSO4·7H2O
1 g (NH4)2SO4
200 mg NaHCO3
300 mg CaCl2·2H2O
0.5 g KCl
420 mg KH2PO4
50 mg NaBr
20 mg SrCl2·6H2O
10 mg Fe(NH4)2(SO4)2·6H2O
Note: Must be autoclaved. - 2x ASW (1 L)
40 g NaCl
6 g MgCl2·6H2O
12 g MgSO4·7H2O
2 g (NH4)2SO4
400 mg NaHCO3
600 mg CaCl2·2H2O
1 g KCl
840 mg KH2PO4
100 mg NaBr
40 mg SrCl2·6H2O
20 mg Fe(NH4)2(SO4)2·6H2O - Trace minerals (1,000x) (1 L)
0.5 g MnSO4·H2O
0.1 g CoCl2·6H2O
0.1 g ZnSO4·7H2O
0.01 g CuSO4·5H2O
0.01 g AlK(SO4)2·12H2O
0.01 g H3BO3
0.01 g Na2MoO4·2H2O - Polysulfides
10 g Na2S·9H2O, 3 g sulfur per 15 ml
Dissolve the mixture using heat
Note: It should be a deep red color when both the sulfur and sodium sulfide have completely dissolved. - 0.8x ASW (1 L)
16 g NaCl
2.4 g MgCl2·6H2O
4.8 g MgSO4·7H2O
800 mg (NH4)2SO4
160 mg NaHCO3
240 mg CaCl2·2H2O
400 mg KCl
336 mg KH2PO4
40 mg NaBr
16 mg SrCl2·6H2O
8 mg Fe(NH4)2(SO4)2·6H2O
Note: Must be autoclaved. - 20 amino acid solution (1 L)
1 g cysteine
1 g glutamic acid
1 g glycine
500 mg arginine
500 mg proline
400 mg asparagine
400 mg histidine
400 mg isoleucine
400 mg leucine
400 mg lysine
400 mg threonine
400 mg tyrosine
300 mg alanine
300 mg methionine
300 mg phenylalanine
300 mg serine
300 mg tryptophan
200 mg aspartic acid
200 mg glutamine
200 mg valine
Acknowledgments
This work was supported by the National Institutes of Health (GM 100329) and Department of Energy (004010-00002) to TJS. The authors declare no conflicts of interest or competing interests.
References
- Aslanidis, C. and de Jong, P. J. (1990). Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res 18(20): 6069-6074.
- Fukui, T., Atomi, H., Kanai, T., Matsumi, R., Fujiwara, S. and Imanaka, T. (2005). Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res 15(3): 352-363.
- Hileman, T. H. and Santangelo, T. J. (2012). Genetics techniques for Thermococcus kodakarensis. Front Microbiol 3: 195.
- Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45(D1): D353-D361.
- Santangelo, T. J., Cubonova, L. and Reeve, J. N. (2008). Shuttle vector expression in Thermococcus kodakaraensis: contributions of cis elements to protein synthesis in a hyperthermophilic archaeon. Appl Environ Microbiol 74(10): 3099-3104.
- Santangelo, T. J., Cubonova, L. and Reeve, J. N. (2010). Thermococcus kodakarensis genetics: TK1827-encoded β-glycosidase, new positive-selection protocol, and targeted and repetitive deletion technology. Appl Environ Microbiol 76(4): 1044-1052.
- Santangelo, T. J., and Reeve, J. N. (2011). Genetic tools and manipulations of the hyperthermophilic heterotrophic archaeon Thermococcus kodakarensis. In: Extremophiles Handbook. Springer 567-582.
- Sato, T., Fukui, T., Atomi, H. and Imanaka, T. (2003). Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J Bacteriol 185(1): 210-220.
- Sato, T., Fukui, T., Atomi, H. and Imanaka, T. (2005). Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl Environ Microbiol 71(7): 3889-3899.
- Untergasser, A., Nijveen, H., Rao, X., Bisseling, T., Geurts, R. and Leunissen, J. A. (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35: W71-74.
- Walker, J. E., Luyties, O. and Santangelo, T. J. (2017). Factor-dependent archaeal transcription termination. Proc Natl Acad Sci U S A 114(33):E6767-E6773.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gehring, A. M., Sanders, T. J. and Santangelo, T. J. J. (2017). Markerless Gene Editing in the Hyperthermophilic Archaeon Thermococcus kodakarensis. Bio-protocol 7(22): e2604. DOI: 10.21769/BioProtoc.2604.
Category
Microbiology > Microbial genetics > DNA > Chromosomal
Microbiology > Microbial genetics > Gene mapping and cloning
Molecular Biology > DNA > Chromosome engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.