- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Uptake Assays to Monitor Anthracyclines Entry into Mammalian Cells

Published: Vol 7, Iss 18, Sep 20, 2017 DOI: 10.21769/BioProtoc.2555 Views: 7956
Reviewed by: Nicoletta CordaniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2016
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Anthracyclines, such as doxorubicin and daunorubicin, are DNA damaging agents that autofluoresce and can be readily detected in cells. Herein, we developed suitable assays to quantify and localize daunorubicin in mammalian cells. These assays can be exploited to identify components that are involved in the uptake of anthracyclines.
Keywords: Influx transportersBackground
The anthracyclines, such as doxorubicin and daunorubicin, act by damaging the DNA and are used for treating various types of cancers including acute myeloid leukemia. When cancer patients are given anthracyclines systemically, there are several factors limiting the amount of the drugs that reach the tumor sites (Chauncey, 2001; Deng et al., 2014; Riganti et al., 2015). In the tumor, the drug activity on the cancer cells is limited by poor drug uptake, excessive drug efflux as well as changes in the cellular targets. For several decades, it remains unclear how these DNA damaging agents enter cancer cells (Aouida et al., 2010; Cesar-Razquin et al., 2015; Zhang et al., 2015). To address this question, we developed three reliable in vitro assays to monitor daunorubicin accumulation into cells. Two of these assays are quantitative and required access to a Fluorescence-Activated Cell Sorting (FACS) caliber and a Fluoroskan instrumentation and the third is semi-quantitative using epifluorescence microscopy. Using these assays, we established that daunorubicin enter into cells in a time- and concentration-dependent manner and that each cell type showed a different rate of uptake, suggesting that an active process is involved in the uptake of anthracyclines (Andreev et al., 2016). We applied these assays and uncovered the organic cation transporter 1 (OCT1) as a key protein for the uptake of daunorubicin into the cells (Andreev et al., 2016). Modulating the level of OCT1 resulting in cells with altered uptake and sensitivity towards daunorubicin (Andreev et al., 2016). These assays provide hints that additional transporters exist to allow uptake of daunorubicin into the cells. We believe that these uptake assays can be exploited further to identify additional factors such as kinases (Tanaka et al., 2004; Ciarimboli and Schlatter, 2005; Zhou et al., 2005; Pelis et al., 2006; Filippo et al., 2011; Sprowl et al., 2016) that influence the rate of daunorubicin uptake. In this protocol, we describe three analyses to monitor the uptake of anthracyclines into cells.
Materials and Reagents
- Mammalian cell culture containers for adherent cells (100 and 60 mm Petri dishes and T-75 flask)
100 mm Petri dishes (SARSTEDT, catalog number: 83.3902 )
60 mm Petri dishes (SARSTEDT, catalog number: 83.3901 )
T-75 flask (Corning, catalog number: 430641 )
Notes:- However, any other Petri dishes or flask suitable for your cell line may work.
- Note that you can grow suspension cells in containers for adherent cells.
- However, any other Petri dishes or flask suitable for your cell line may work.
- Eppendorf tubes
- FACS tubes that will adapt to the flow cytometer:
Falcon 5 ml polystyrene round-bottom tube (Corning, Falcon®, catalog number: 352058 ) - Microscope slides (UltiDent Scientific, catalog number: 170-7107A )
Note: Any microscope slide that fit on your microscope can be used. - Cover glass (UltiDent Scientific, catalog number: 170-C1818 )
Note: Any cover glass can be used. The best cover glasses are the #1.5, however #1.0 or #0 may be used if high quality images are not required (e.g., confocal microscopy). - Black clear flat bottom 96-well plates (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 165305 )
- Mammalian adherent cells, for example, HeLa, HEK293 and TOV112D
Note: Suspension cells such as HL60 and K562 can be used, although the protocol would need to be adjusted. - Required complete culture media
- DMEM (WISENT, catalog number: 319-005-EL )
- 1x RPMI 1640 (WISENT, catalog number: 350-000-CL )
- Ovarian Surface Epithelial (OSE) (WISENT, catalog number: 316-030-CL )
- Fetal bovine serum (FBS) (WISENT, catalog number: 095150 )
Note: It is usually used at 10%, but some cell lines will require different concentrations. See with your cell line supplier. - Penicillin/streptomycin (WISENT, catalog number: 450-201-EL )
Note: It is usually supplied as a 100x stock and therefore must be diluted 1:100 in DMEM and RPMI1640. - Gentamycin Sulfate 50 mg/ml Solution (WISENT, catalog number: 450-135-XL ) supplied at 1,000x and must be diluted 1:1,000 in the media
- Amphotericin B (Sigma-Aldrich, catalog number: A4888 ) used at 0.5 μg/ml in OSE
Note: We usually prepare a stock solution of 250 μg/ml and add 1 ml of to 500 ml of OSE.
- DMEM (WISENT, catalog number: 319-005-EL )
- Trypsin (0.05% or 0.25% in PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 25200072 )
- Daunorubicin
Notes: - 4% paraformaldehyde (PFA) (Sigma-Aldrich, catalog number: P6148 ) solution in PBS
- Mounting medium with DAPI (Vectashield, Vector Laboratories, catalog number: H-1200 )
Note: This component may be replaced by 50% glycerol (Bio Basic, catalog number: GB0232 ) containing 1 to 10 μg/ml Hoechst 33342 (Thermo Fisher Scientific, InvitrogenTM, catalog number: H1399 ). - Nail polish
- Phosphate buffer saline (PBS) (see Recipes)
Sodium chloride (NaCl) (WISENT, catalog number: 600-082-IK )
Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P4504 )
Sodium phosphate dibasic (Na2HPO4) (Bio Basic, catalog number: S0404 )
Potassium phosphate monobasic (KH2PO4) (Bio Basic, catalog number: PB0445 ) - Uptake buffer (see Recipes)
Sodium chloride (NaCl) (WISENT, catalog number: 600-082-IK )
HEPES pH 7.4 (Bio Basic, catalog number: HB0265 )
Glucose (WISENT, catalog number: 600-350-IK )
Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P4504 )
Potassium phosphate monobasic (KH2PO4) (Bio Basic, catalog number: PB0445 )
Calcium chloride (CaCl2) (Fisher Scientific, catalog number: C79-500 )
Magnesium sulfate (MgSO4) (Bio Basic, catalog number: MN1988 )
Equipment
- Pipettes (any pipettes will do)
- Incubator (any incubator is fine if it maintains 5% CO2 and moisture)
- pH meter (any pH meter will work)
- Eppendorf centrifuge (any centrifuge that can adapt 1.5 ml microcentrifuge tube)
- Hemocytometer (any hemocytometer will do)
- Flow cytometer (FACS)
Note: This protocol is using a FACS Calibur from Becton-Dickson (BD, model: FACS CaliburTM ). - Epifluorescence microscope (Olympus, model: BX53 )
Note: We use an Olympus BX53 , but any epifluorescence microscope with the required filter sets may be used (see in Procedure D for the required filters). - Fluoroskan Ascent (Thermo Fisher Scientific, Thermo ScientificTM, model: Fluoroskan AscentTM )
Software
- ImageJ
Note: We used the one at the National institute of Health website; https://imagej.nih.gov/ij/. - CellQuest Pro either version 4.0.1 © 1994-2001 BDB or 5.2.1 © 1994-2005 BD
Procedure
- Preparation of the cells
- Grow the cells of interest in the recommended culture media at 37 °C, 5% CO2, 95% air.
- The day before the experiment, remove media from the cells and wash with PBS (see Recipes). Add appropriate volume of trypsin (1-2 ml for 100 mm Petri dish or T-75 flask). Incubate at 37 °C for 2 to 15 min.
Note: The incubation time varies between cell lines. Incubate until you can see the cells detaching from the surface. - Add complete media to the cells to inhibit the trypsin.
Note: The volume of media to add to the cells depends on the number of samples to be created. Keep in mind that you have to divide this volume by the number of samples you want to create. - Pipette the cells into 60 mm Petri dishes with a total of about 5 to 6 ml of media.
Notes:- The number of plates needed depends on the kind of experiment you want to perform. You always need at least one Petri plate for negative control, but it is better to have all samples in triplicate. You will need three plates for each time point or three plates for each daunorubicin concentrations to be tested.
- You must seed your cells so the confluence on the day of the experiment is below 50% as higher confluence will artificially reduce daunorubicin uptake.
- The number of plates needed depends on the kind of experiment you want to perform. You always need at least one Petri plate for negative control, but it is better to have all samples in triplicate. You will need three plates for each time point or three plates for each daunorubicin concentrations to be tested.
- Incubate cells at 37 °C, 5% CO2, 95% air.
- Grow the cells of interest in the recommended culture media at 37 °C, 5% CO2, 95% air.
- Treatment of the cells with daunorubicin
- Remove the culture media from the cells.
Note: Cells can be washed, but this does not seem to affect the overall results. However, if cells were treated with additional compounds prior to daunorubicin treatment, a washing step with UB should be added. - Add 5 ml of UB to the cells.
- Incubate at 37 °C, 5% CO2, 95% air for at least 5 min.
- Prepare a daunorubicin dilution of 255 µM in UB by diluting the stock concentration of 8.87 mM by 35-fold.
- Add 100 µl of 255 µM daunorubicin to the 5 ml of UB to have a final concentration of 5 μM.
Notes:- To test different concentrations of daunorubicin, add the required volume of diluted daunorubicin keeping in mind to adjust the volume of UB added to the cells before. Trying various concentrations may be a good thing in order to determine the best concentration for your experiment. The reference concentration should be 5 µM.
- Keep in mind that you will need an untreated cell sample for all the manipulations.
- To test different concentrations of daunorubicin, add the required volume of diluted daunorubicin keeping in mind to adjust the volume of UB added to the cells before. Trying various concentrations may be a good thing in order to determine the best concentration for your experiment. The reference concentration should be 5 µM.
- Incubate the cells at 37 °C, 5% CO2, 95% air for the required time.
Note: Daunorubicin uptake can be detected after a few minutes, but longer exposure time may increase the difference between samples. Therefore, a time-dependent curve may be necessary in order to determine the best time point for your experiment. An exposure time between 30 to 60 min would be the best starting point. - Once the incubation at 37 °C is finished, depending on the cell type they may start to detach from the plate. If this is the case, collect the cells by up and down pipetting to detach all the cells and put them in Eppendorf tubes. Pellet the cells at 5,000 x g for 1 min using any standard Eppendorf centrifuge and remove the supernatant. Wash the cells with 1 ml of cold PBS.
Note: If the cells did not detach from the plate, remove the UB containing the daunorubicin and wash the cells with cold PBS. Add 0.5 ml of trypsin and wait for the cells to detach. Add 0.5 ml of cold PBS and collect all the cells in an Eppendorf tube and centrifuge at 5,000 x g for 1 min. Remove supernatant. - To the cell pellets, add 100 µl of 4% PFA and incubate for 10 min at room temperature or 60 min at 4 °C.
- Centrifuge 5,000 x g for 1 min and remove the supernatant.
- Add 100 µl of PBS.
Note: From this point, you can go to either Procedure C (FACS analysis), Procedure D (Epifluorescence Microscopy) or Procedure E (Fluoroskan analysis).
- Remove the culture media from the cells.
- FACS analysis of the treated cells (need roughly 100,000 cells for the analysis)
- Take one FACS tube for each sample you have and put 250-500 µl of PBS in each tube.
Note: The amount of PBS you put in the tube depends on the number of cells you have. More PBS you put, more time it will take to reach the required number of events on the FACS. However, you need sufficient volume to make your adjustment on the machine. - Add the cells prepared in Procedure B to the PBS in the FACS tube.
Note: You can always keep some cell suspension to perform epifluorescence microscopy and Fluoroskan analyses as in Procedure D and Procedure E, respectively, but this would depend on the size of the cell pellets in Procedure B. Keep in mind that it will take longer to read 10,000 events if there are less cells in the FACS tube. - Turn on the FACS and the computer and make sure the valve is closed so the FACS Flow tank is under pressure.
The tanks are in the ‘drawer’ of the machines. There are two tanks, the right one is the FACS Flow, the left one is for the wastes. The valve is at the back between the tanks. - Open the software of the FACS (here we are using CellQuest Pro).
- Go to ‘Acquire’ tab and click on ‘Connect to cytometer’ (Figure 1).
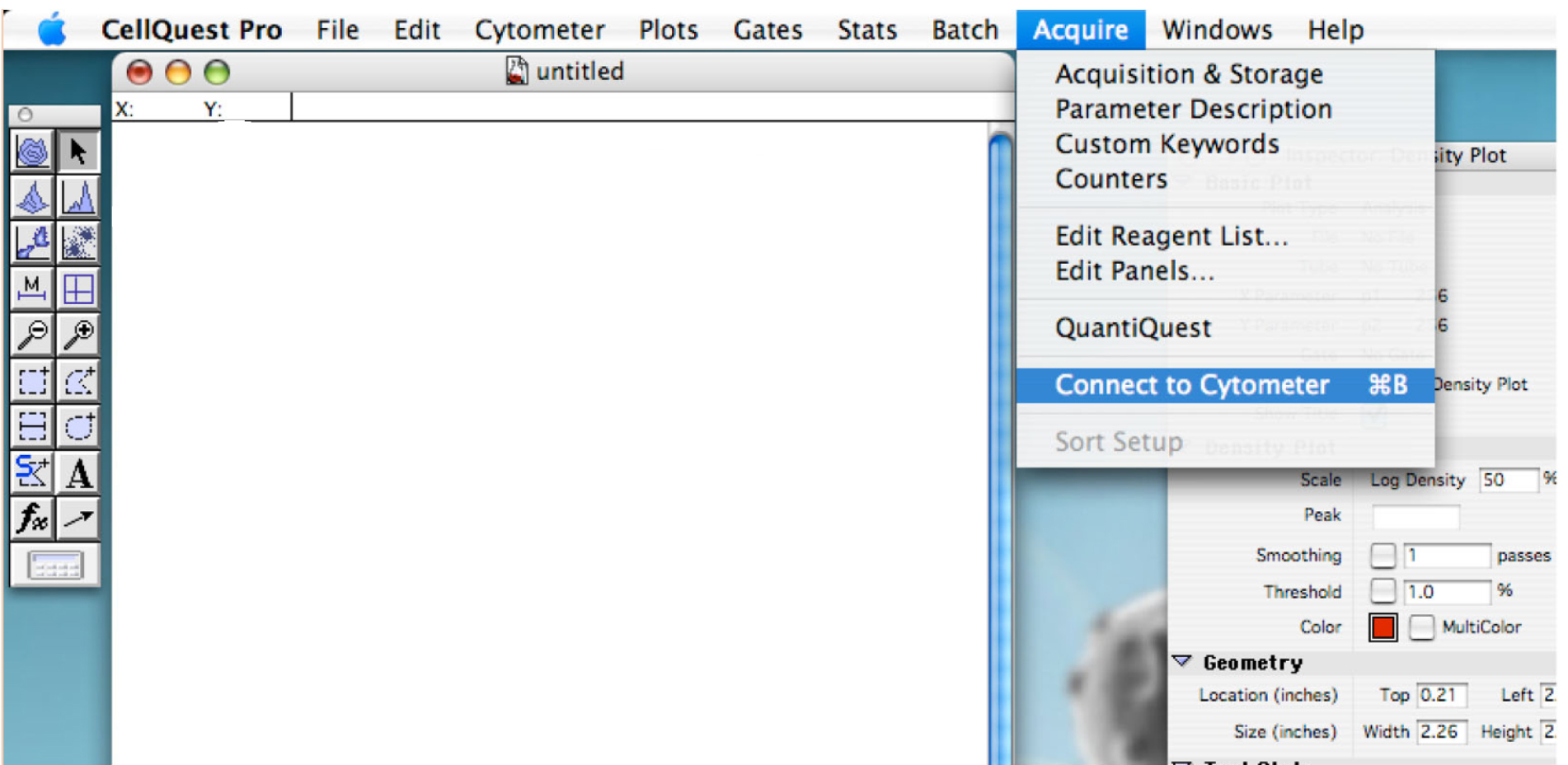
Figure 1. Showing the display to connect the cytometer to the computer - Enter the name for each needed detector. Use P4 (FL2 585/42) for daunorubicin detection (Figure 2).
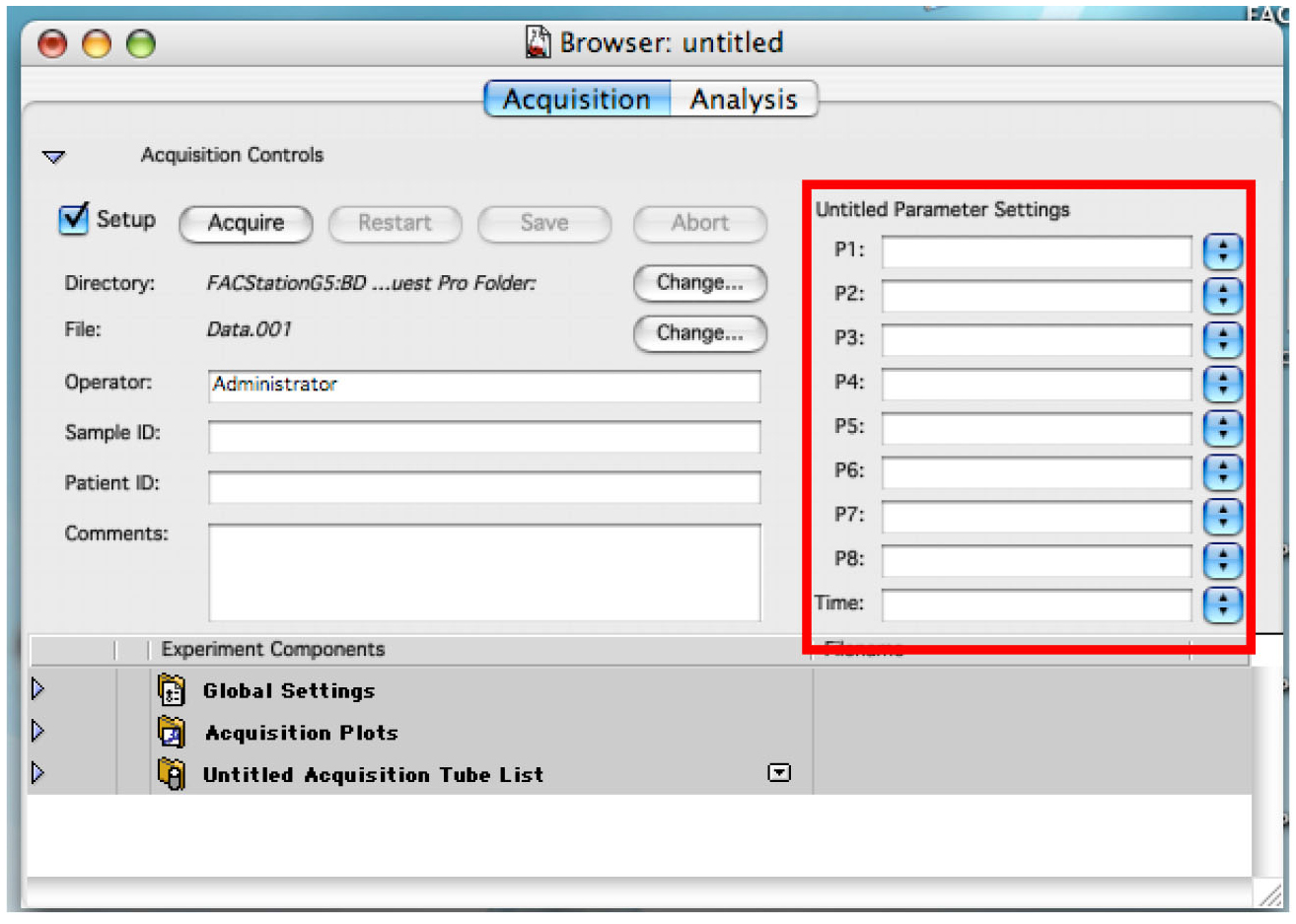
Figure 2. Displaying the Browser. Enter names for each used detector. - Choose the folder where you want to save your data by clicking on the first ‘Change’ (Figure 3).
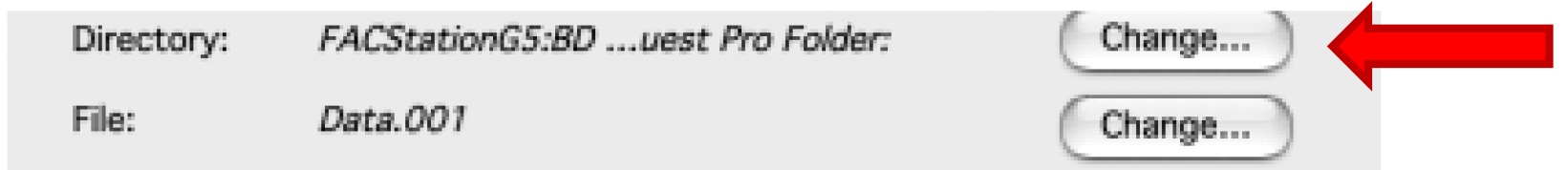
Figure 3. Displaying within the Browser, the Directory. Selection of the folder that will contain all the file of the experiment. - Draw a dot plot or a density plot and change the ‘Plot Type’ to acquire˃˃analysis (Figure 4).
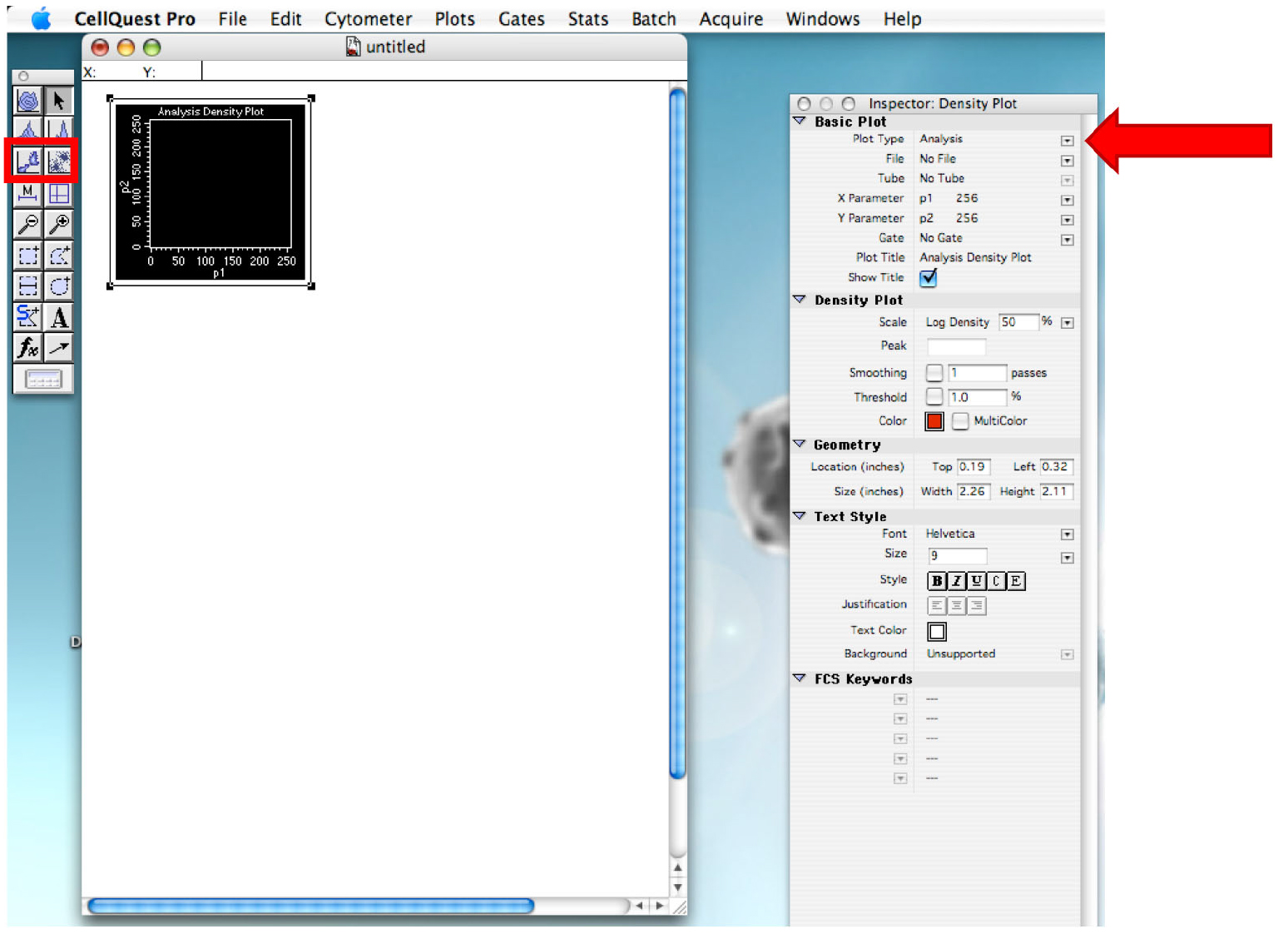
Figure 4. Plot creation. Make sure to change the plot type. - Click on ‘Edit’ tab and ‘Duplicate’ to create another graph. On the second graph, change the x axis name to daunorubicin.
Note: This is like doing ‘copy/paste’, but it doesn’t work to duplicate graph in CellQuest Pro and this is why one must use ‘duplicate’ function. Graphs could also be created one by one, but this requires changing the ‘plot type’ each time. - In the ‘Cytometer’ tab, click on ‘Detector/Amps’. Change the fluorescent one to ‘Log’ mode (Figure 5).
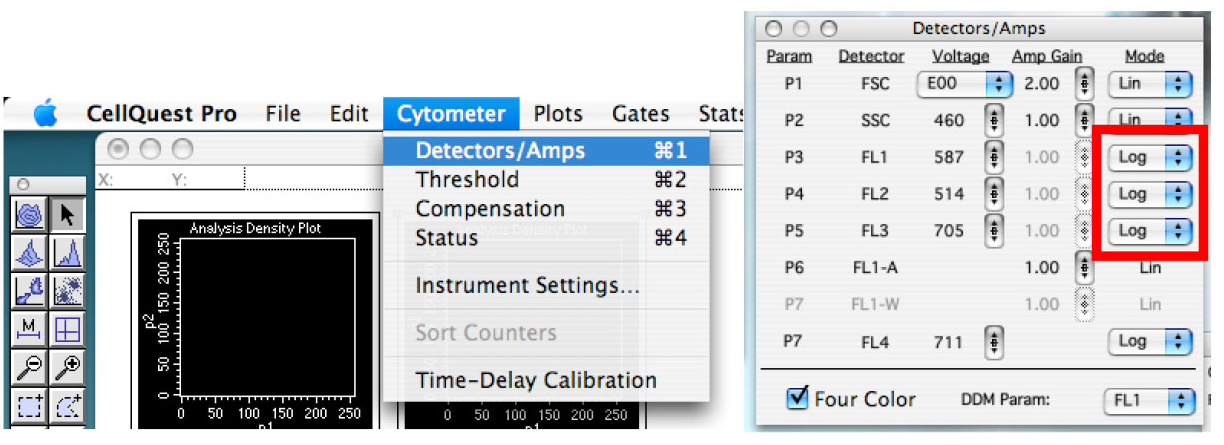
Figure 5. Showing the detector controls. Change the scale type and adjust voltage of detectors. - Click on the quadrant tool and create quadrants in the daunorubicin graph placing the vertical line around 10 and the horizontal line around the middle of the graph (Figure 6).
These will constitute thresholds. You can change these settings according to your sample if required.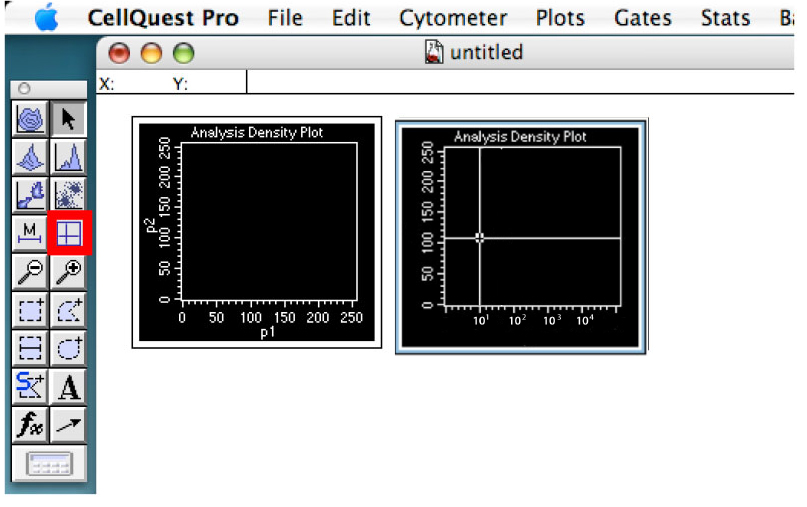
Figure 6. Showing the quadrant. The thresholds of your experiment. - Click on the ‘Stats’ tab and ‘Quadrant Stats’ (Figure 7).
You can edit the quadrant stats if you go back in the ‘Stats’ tab (Figure 7).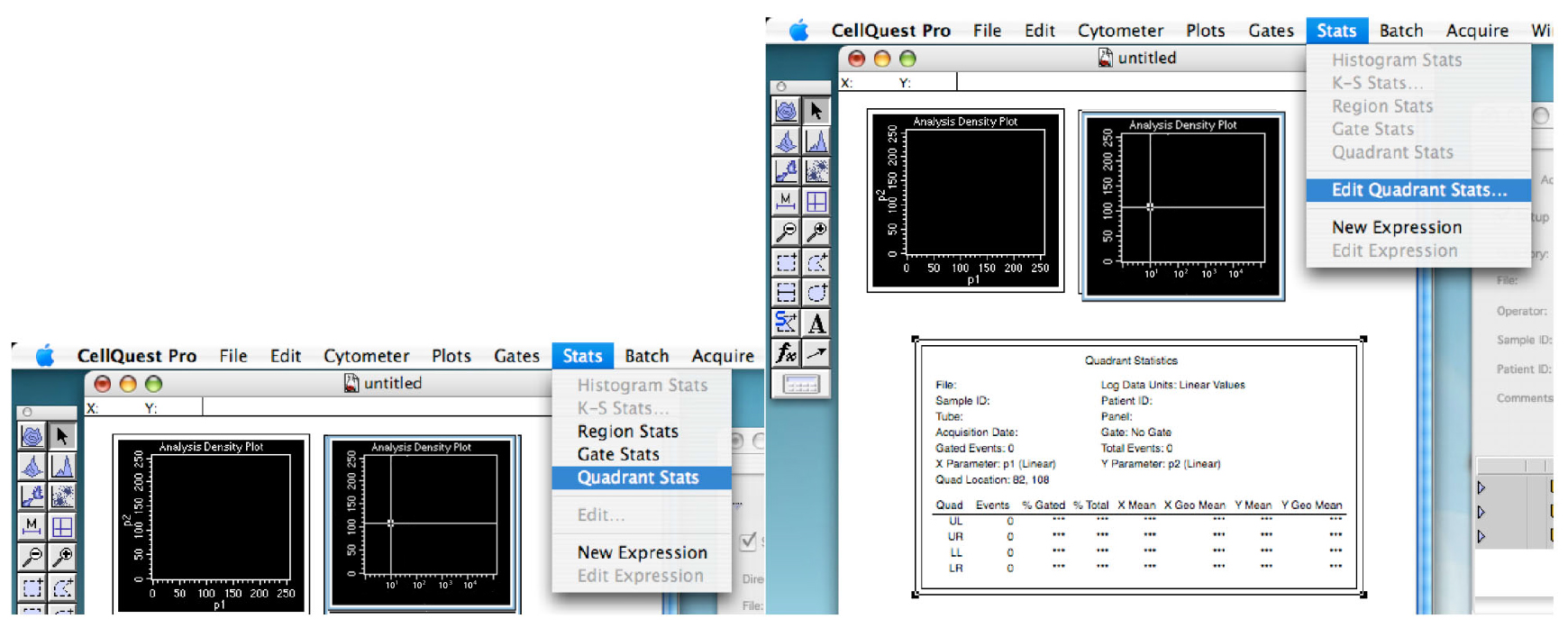
Figure 7. Display and edit quadrant stats - Remove any tube from the machine and press ‘Prime’. Once the light of the button is turned off, press again.
- Place one of your negative control tubes on the machine and press ‘Run’.
- Ensure the ‘Setup’ check box is checked and click on ‘Acquire’ (Figure 8).
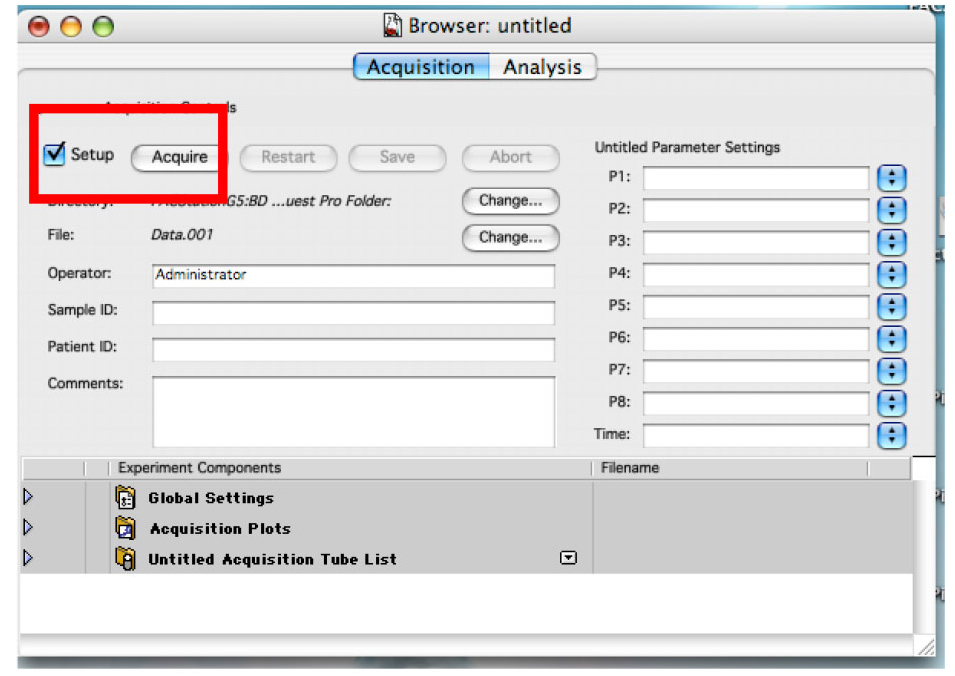
Figure 8. The Browser in ‘Setup’ mode. This allows you to analyze cells without saving any data. - Change the voltage of the detectors to have almost all the cells in the lower left quadrant. (Figure 9)
Notes:- Using SSC as y axis, place the horizontal line of the quadrants such as to exclude cells with a very high SSC value which have high chances of being dead cells. Thus, the upper quadrants will exclude the dead cells.
- Another option to exclude dead cells from the analysis is to create a gate on the graph displaying FSC and SSC of the cells (P1 and P2). Dead cells will have a very low FSC and a high SSC. To do so, on the FSC-SSC graph, use the gate tools to select the living cell population. In the menu of the daunorubicin graph, go to ‘Gate’ and select ‘R1’ or the appropriate gate.
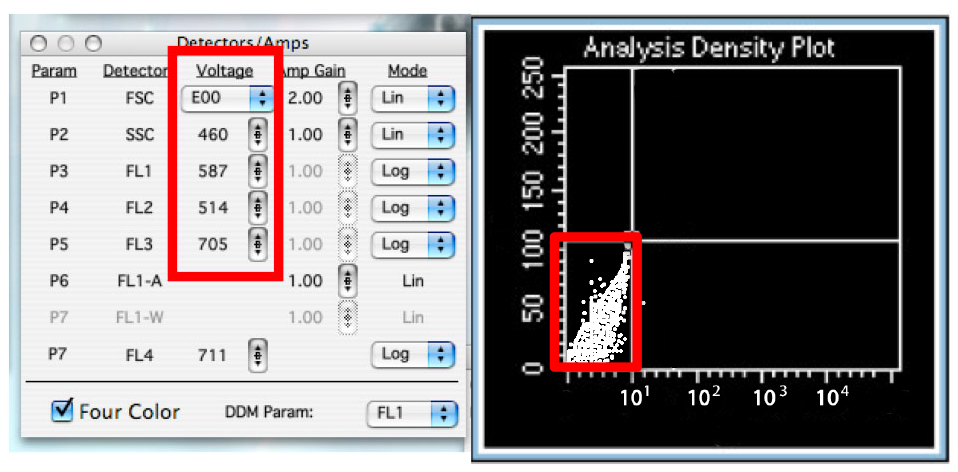
Figure 9. Adjust detectors voltage - Using SSC as y axis, place the horizontal line of the quadrants such as to exclude cells with a very high SSC value which have high chances of being dead cells. Thus, the upper quadrants will exclude the dead cells.
- Click on the ‘Set Up’ check box to remove the check and write the name of your first sample in the sample text box (Figure 10).
You can also use the patient text box to write part of the name that will not change often. As an example, if you are doing time dependent curve with two cell lines, write the cell line’s name in the patient text box and the time point in the sample text box (Figure 10).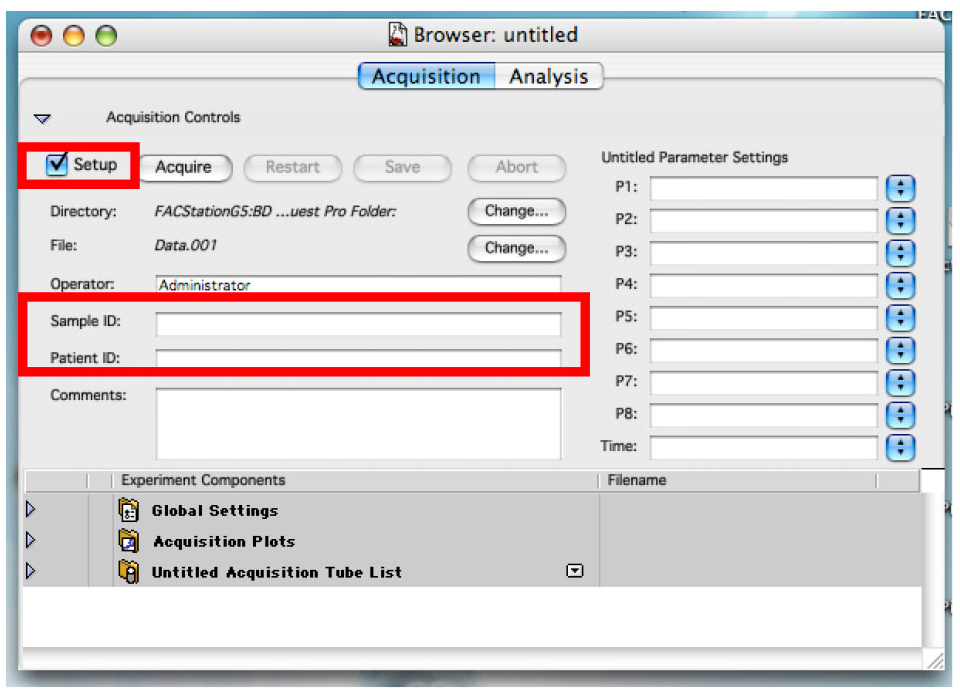
Figure 10. Preparing the acquisition - Place the sample to be analyzed on the machine and click on ‘Acquire’ (Figure 11).
You can see the number of events per second and the total number of events by displaying the ‘Counters’ window in the ‘Acquire’ tab. The speed of the machine can be changed from slow to medium to high directly on the machine. Having 1,000 events per second is the best.
Figure 11. Acquisition - Change your tube and repeat the acquisition for all your samples.
- Once you have analyzed all your samples, you can display them in the graph by clicking on the graph and then, in the ‘Plot’ tab you can change data displayed (Figure 12).

Figure 12. Navigation between samples - To analyze the data, multiply the mean daunorubicin fluorescence in the lower right quadrant by the percentage of cells in that quadrant displayed in the quadrant stats window (Figure 13).
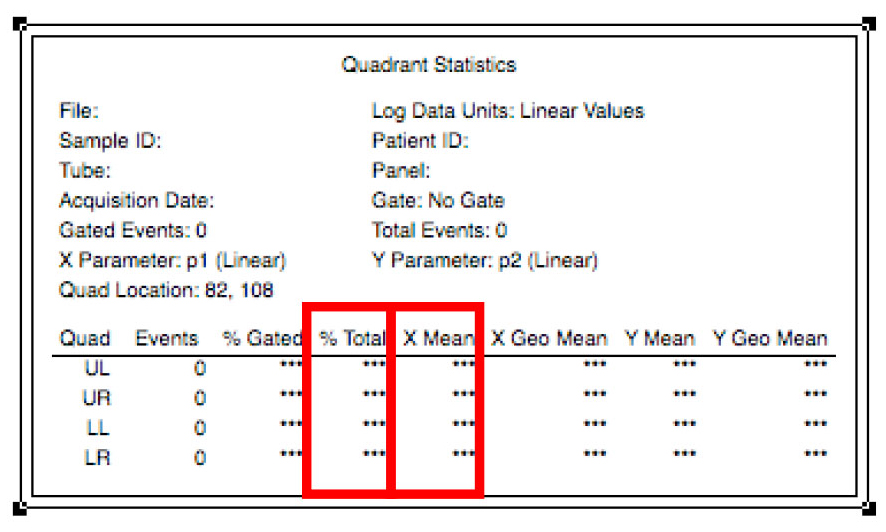
Figure 13. Calculation of the average fluorescence of the sample
Note: If you are using cell line transfected with a fluorescent marker, for example, studying the effect of overexpression of a particular protein tagged with GFP, create another graph and change the x axis of that graph for P3 (FL1 530/30). Create a gate using the gating tools to select only the GFP positive cells using a non-transfected cell sample as negative control to set the threshold. Next, within the menu of the graph that displays the daunorubicin fluorescence, go to Gate and select R1. This way, the graph displaying the daunorubicin will only display GFP positive cells. This can be very useful if you are doing transient transfection. However, you might need to increase the number of events to get enough GFP positive cells in the daunorubicin graph. To do so, go in the Acquire tab and click on acquisition and storage. There you can set the even count. There could be GFP leak on the FL2 detector, as such using P5 (FL3) may be an option. You can also use the compensation menu in the cytometer tab to reduce the effect of the GFP on the FL2 and the effect of the daunorubicin on the F1.
- Take one FACS tube for each sample you have and put 250-500 µl of PBS in each tube.
- Epifluorescence microscopy
- Put 3 μl of mounting medium with DAPI on a microscope slide.
Note: Mounting medium with DAPI can be replaced by 50% glycerol solution containing 1 to 10 μg/ml of Hoechst 33342. - Add 2 μl of cell suspension from Procedure B.
- Put a cover slip on the sample and fix it with nail polish.
Note: Slides can be stored at 4 °C in the dark for one to two weeks. - Visualize the cell with an epifluorescence microscope using DAPI filter to visualize nucleus and Red filter (Texas RED) to visualize daunorubicin at 60x or 100x.
Note: The protocol here describes the procedure for an Olympus B53 upright epifluorescence microscope working on Cellsens.- DAPI filter is working also for staining with Hoechst 33342.
- To visualize daunorubicin you must excite with a wavelength going from 480 nm to 560 nm. Excitation through a FITC or Texas RED filter should work. The emission of daunorubicin can be detected from 575 nm to 680 nm (Texas RED/mCHerry filter). Using a Texas RED filter set would be the best, otherwise different filters for emission and excitation could be required.
- DAPI filter is working also for staining with Hoechst 33342.
- Using a control sample, set the light intensity and the exposure time so the daunorubicin is clearly visible, but not saturated (Figure 14).
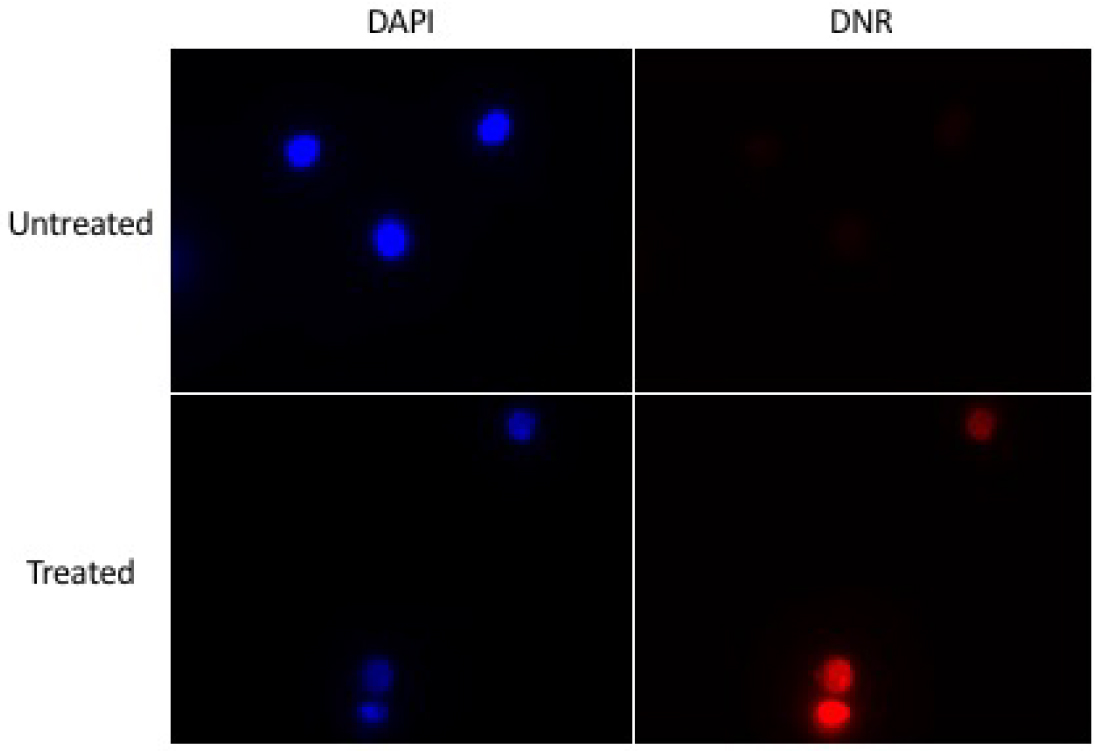
Figure 14. Example of images to be obtained - To adjust the light intensity, use lamp device connected to the microscope.
- The adjustment of the exposure time is done on the computer in the ‘exposure window’ (Figure 15).
To use this method in a quantitative way you must not change these parameters for the daunorubicin visualization.
Figure 15. Adjusting the exposure time - Adjust these parameters (light intensity and exposure time) also for nucleus.
These settings can be changed from one sample to another unless you want to study the chromatin itself. The staining of the chromatin by DAPI or Hoechst can be compromised by high amount of DNR onto the DNA. - If using GFP or another color, set the light intensity and exposure time for this color as well.
- Take pictures of all samples for all colors. To do so, you must click on ‘Snap’ (Figure 16).
Figure 16. Camera controls. Snap to take a picture. - You can navigate between the pictures you took by clicking on the tabs above the pictures (Figure 17).
Notes:- The ‘live’ tab allows you to see live what is on the microscope. To display it, if it is not, click on the live button.
- Important Note: Microscopes have ‘modes’ so they display through the ocular, the camera or both. When you take picture, make sure the ‘mode’ is one that is using the camera. On Olympus B53, this can be by pulling or pushing a metal stick on the right side of the oculars.
- Some microscopes such as ZEISS Z2 use special software that can also serve to edit the pictures to create TIFF files with colors and a merge pictures.
Figure 17. Navigation between the pictures - The ‘live’ tab allows you to see live what is on the microscope. To display it, if it is not, click on the live button.
- Save all the images which can then be processed with ImageJ.
- Open the ImageJ software and click on file > to open the images. Open all the colors of a single sample (Figure 18).
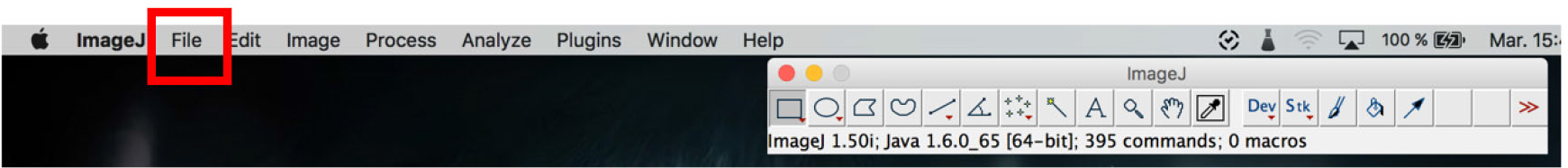
Figure 18. Open pictures in ‘File’ - Click on the picture, then go to the ‘image’ > channels tool (Figure 19).
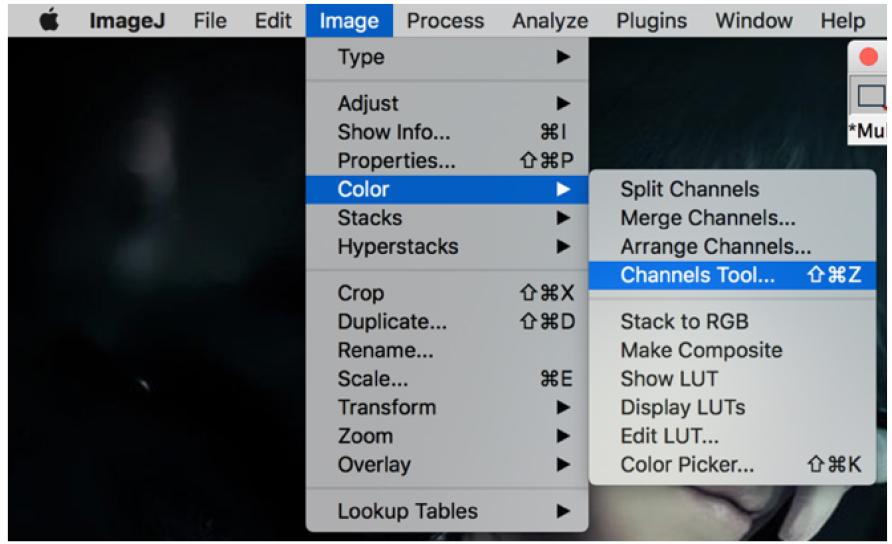
Figure 19. Open the ‘Channels Tool’ - On the channel window, click on ‘more’ and then click on the required color for your selected picture. Apply the colors to all pictures (Figure 20).
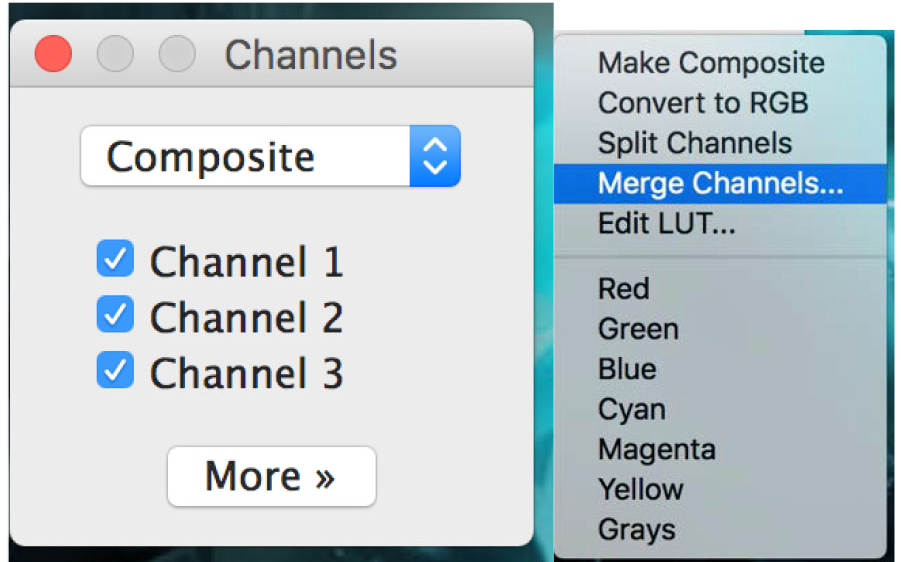
Figure 20. Apply the colors to your pictures - Click on ‘Merge Channels’ and select the required picture for each color. Click ‘OK’ (Figure 21).
If you do not want the software to close all your opened pictures, make sure the ‘Keep source images’ check box is checked (Figure 21). - You can now ‘Save as’ all pictures as desired file type: TIFF, JPEG, etc.
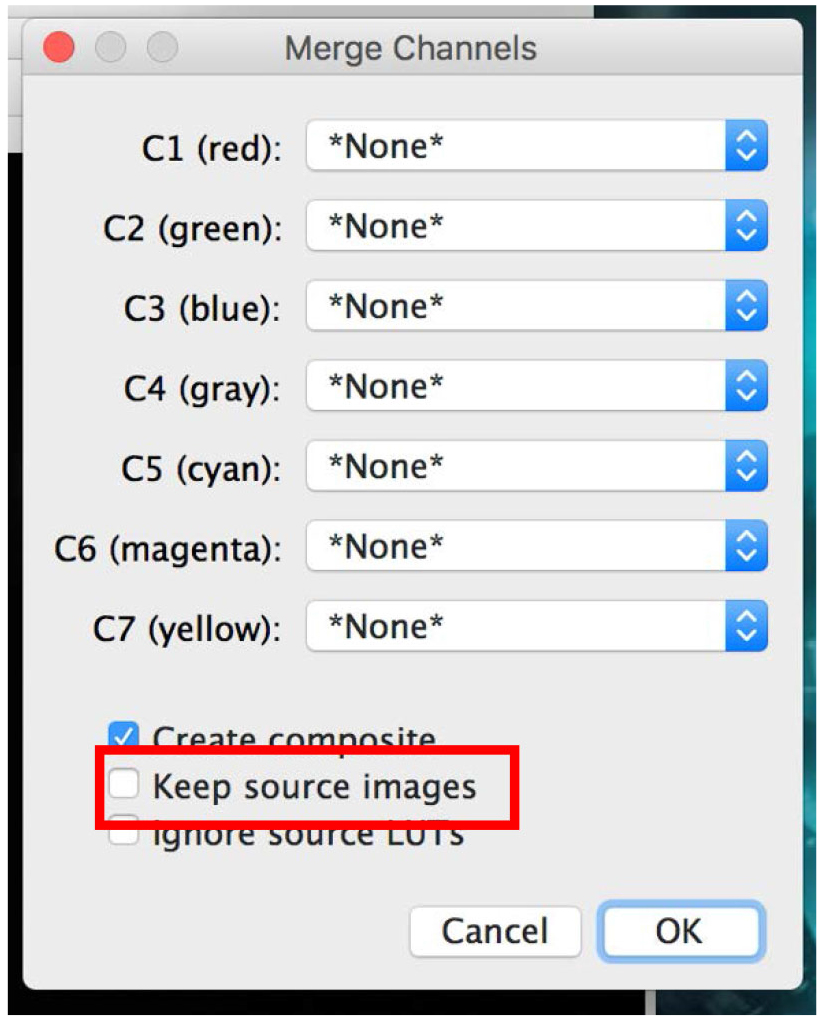
Figure 21. Merge your images
- Put 3 μl of mounting medium with DAPI on a microscope slide.
- Fluoroskan analysis
- Dilute the cell suspension from Procedure B 1:10 to 1:100 depending on the cell density.
- Put 10 μl of the dilution on a hemocytometer and count the number of cells in the 25 central squares and multiply the number by 20,000.
- Then multiply the number obtained by the dilution factor to obtain the number of cells per ml in the cell suspension.
Note: Do this for each sample to be analyzed. - In a black clear flat bottom 96-well plate, put 6,000 to 20,000 cells and complete the volume in each well to 100 μl with PBS. Each well must have the same amount of cells.
Note: It is recommended to do analyze each sample in triplicate. - Put the plate on the Fluoroskan plate reader.
Note: Make sure to remove the cover of the 96-well plate. - Click on ‘Measure1’ (Figure 22).
Click on ‘Measure1’ (Figure 22).
Figure 22. Open the ‘Measure1’ window - Select the wavelength pair 544/590 (Figure 23).
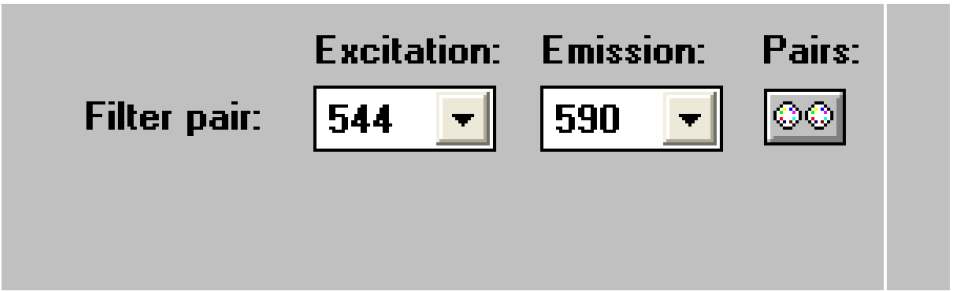
Figure 23. Select the right wavelength pair - Click on ‘Shake1’ (Figure 24).
Figure 24. Open the ‘Shake1’ window - Set a time of 30 sec and a speed that is no more than 900 rpm (Figure 25).
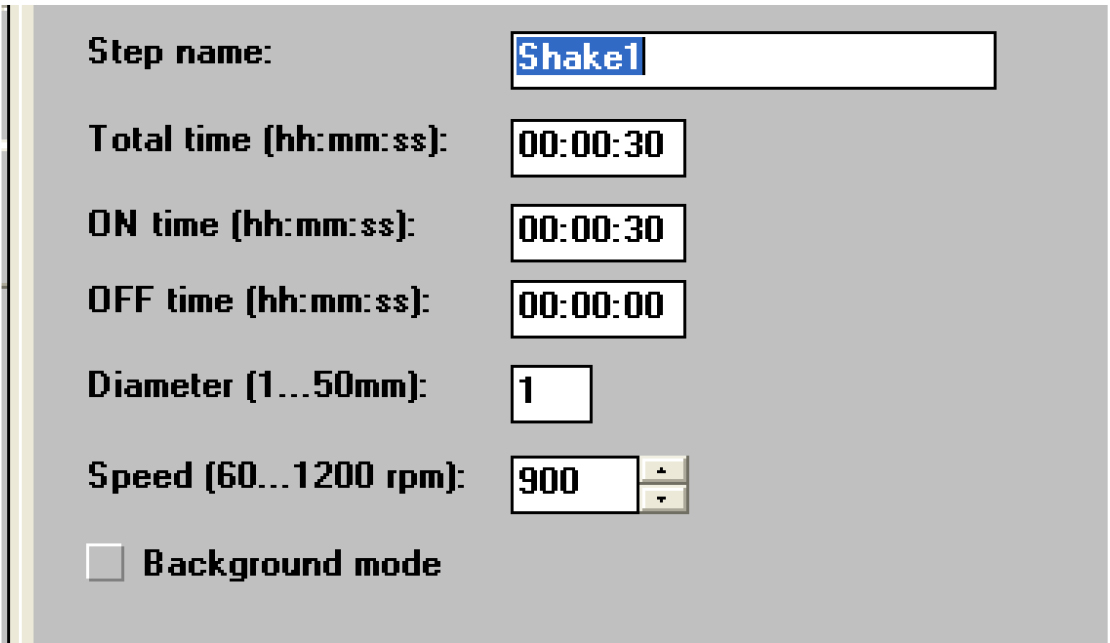
Figure 25. Settings for shaking the samples. Not too fast! - Click on ‘General’ (Figure 26).
Figure 26. Open the ‘General’ window - Click on ‘area definition’ tab and select the wells to read by dragging the mouse over them (Figure 27).
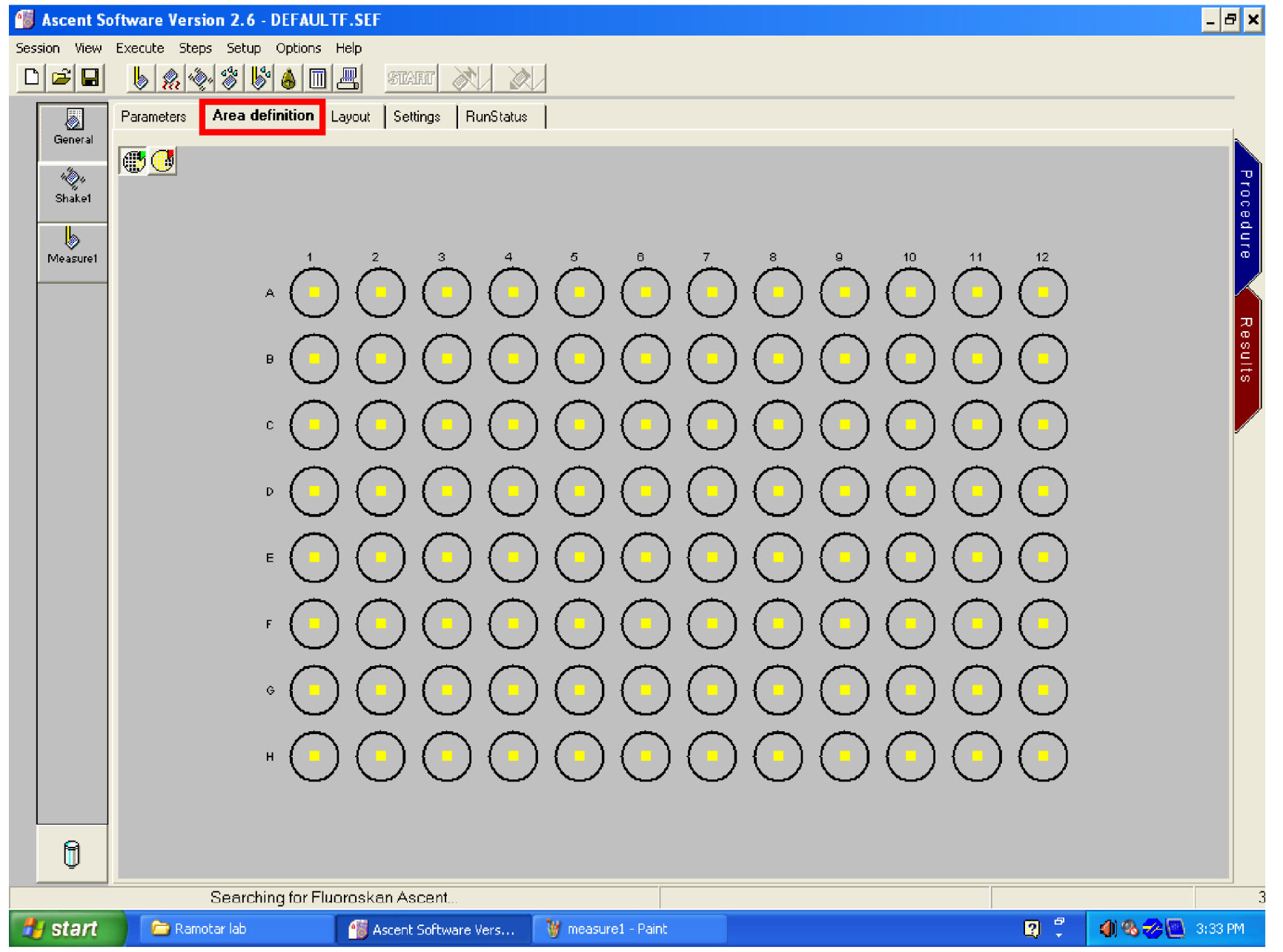
Figure 27. Area setting
To remove wells, make sure the red well icon is click. To add wells, make sure the green well icon is clicked (Figure 28).
Figure 28. Area setting controls - Click on ‘Layout’ (Figure 29). Clear any layout on the screen by dragging the mouse over the wells then click on ‘Clear’ (Figure 29). Select ‘BLANK’ in ‘Type’ (Figure 29) then select the wells containing your untreated samples. Click on ‘Apply’ (Figure 29).
- Change ‘Type’ to ‘SAMPLE’ and select all the wells containing treated cells. Click on ‘Apply’ (Figure 29).
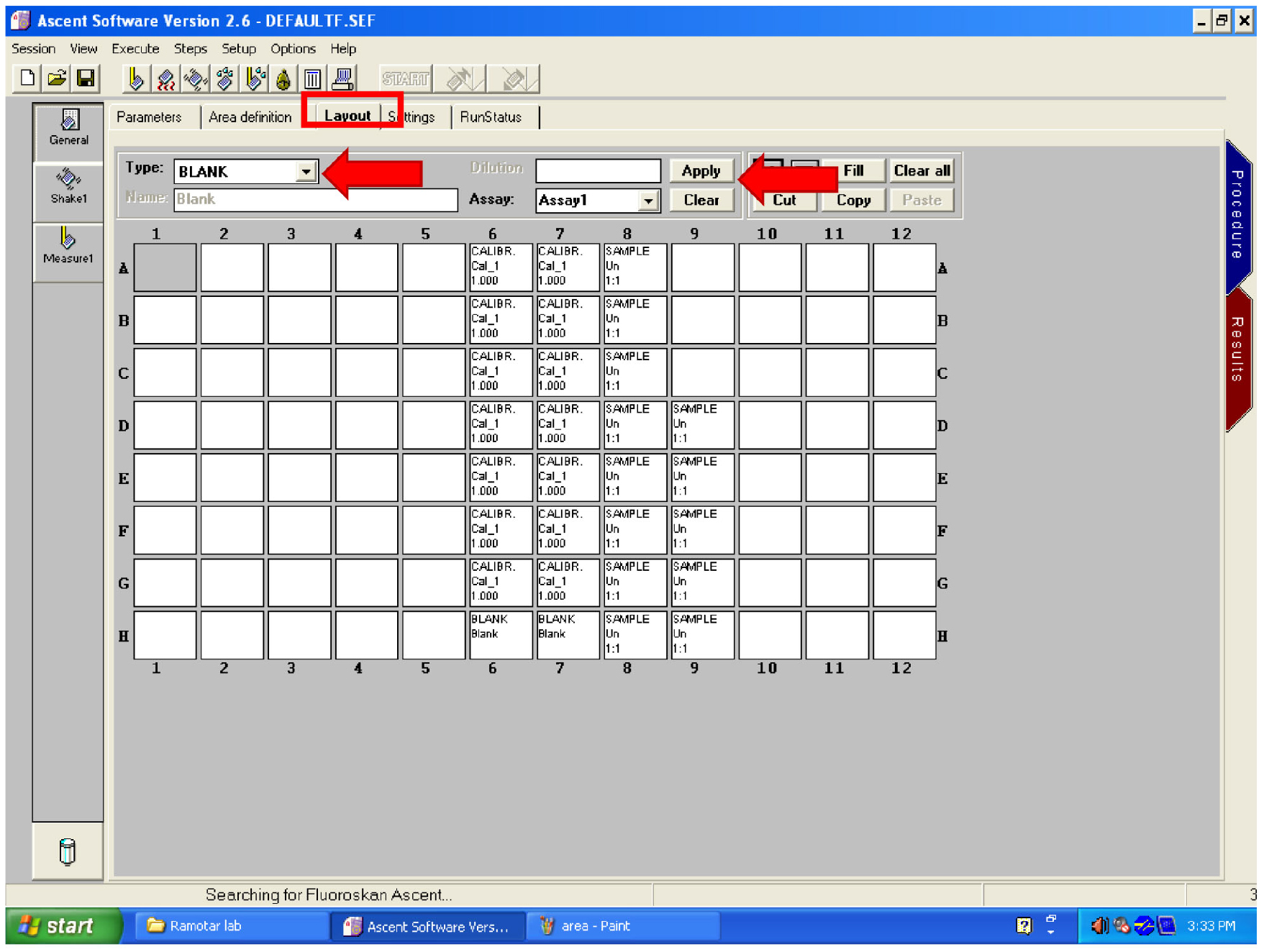
Figure 29. Set the layout - Click on the START button (Figure 30).
Figure 30. Read your samples
- Dilute the cell suspension from Procedure B 1:10 to 1:100 depending on the cell density.
Data analysis
For Procedure C (FACS analysis) and Procedure E (Fluoroskan analysis), the calculations are straightforward which involve taking the values obtained at the end of each procedure and determine the average and standard deviation. Using a reference sample and express the others as a percentage is a good idea especially if there is a need to compare different experiments (Figure 31).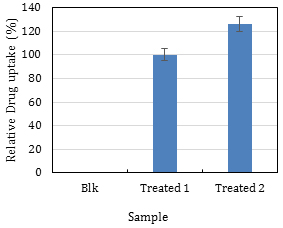
Figure 31. Plotting the FACS results. Sample ‘Treated 1’ was used as the reference sample and adjusted to 100%, and sample ‘Treated 2’ was expressed relative to this reference. This graph shows that sample ‘Treated 2’ takes up 20% more drug than the reference sample.
Note: The blank sample in Figure 31 does not need to be absolutely zero, but its value must be negligible. Adjust the voltage of the DNR detector so that > 95% of the Blank cells are on the left of the line (Figure 9). It is recommended to subtract the values of the blank from all other values (e.g., zero time point).
Recipes
- Phosphate buffer saline (PBS) pH 7.4 (1 L)
8 g NaCl
0.2 g mM KCl
1.44 g mM Na2HPO4
0.24 g mM KH2PO4 - Uptake buffer pH 7.4 (100 ml)
12.5 ml 1 M NaCl
2.0 ml 0.5 M HEPES pH 7.4
0.6 ml 1 M glucose
0.5 ml 1 M KCl
0.6 ml 0.2 M KH2PO4
0.1 1 M CaCl2
0.6 ml 0.2 M MgSO4
Acknowledgments
This work was funded by the research grant MOP-93573 to D.R. from the Canadian Institute of Health Research. We thank Dr. Mikhail Sergeev for providing technical guidance with the microscopes and Martine Dupuis for technical guidance with the FACS. A brief version of this protocol was previously described (Andreev et al., 2016).
References
- Andreev, E., Brosseau, N., Carmona, E., Mes-Masson, A. M. and Ramotar, D. (2016). The human organic cation transporter OCT1 mediates high affinity uptake of the anticancer drug daunorubicin. Sci Rep 6: 20508.
- Aouida, M., Poulin, R. and Ramotar, D. (2010). The human carnitine transporter SLC22A16 mediates high affinity uptake of the anticancer polyamine analogue bleomycin-A5. J Biol Chem 285(9): 6275-6284.
- Cesar-Razquin, A., Snijder, B., Frappier-Brinton, T., Isserlin, R., Gyimesi, G., Bai, X., Reithmeier, R. A., Hepworth, D., Hediger, M. A., Edwards, A. M. and Superti-Furga, G. (2015). A call for systematic research on solute carriers. Cell 162(3): 478-87.
- Chauncey, T. R. (2001). Drug resistance mechanisms in acute leukemia. Curr Opin Oncol 13(1): 21-26.
- Ciarimboli, G. and Schlatter, E. (2005). Regulation of organic cation transport. Pflugers Arch 449(5): 423-441.
- Deng, S., Yan, T., Jendrny, C., Nemecek, A., Vincetic, M., Godtel-Armbrust, U. and Wojnowski, L. (2014). Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer 14: 842.
- Filippo, C. A., Ardon, O. and Longo, N. (2011). Glycosylation of the OCTN2 carnitine transporter: study of natural mutations identified in patients with primary carnitine deficiency. Biochim Biophys Acta 1812(3): 312-320.
- Pelis, R. M., Suhre, W. M. and Wright, S. H. (2006). Functional influence of N-glycosylation in OCT2-mediated tetraethylammonium transport. Am J Physiol Renal Physiol 290(5): F1118-1126.
- Riganti, C., Gazzano, E., Gulino, G. R., Volante, M., Ghigo, D. and Kopecka, J. (2015). Two repeated low doses of doxorubicin are more effective than a single high dose against tumors overexpressing P-glycoprotein. Cancer Lett 360(2): 219-226.
- Sprowl, J. A., Ong, S. S., Gibson, A. A., Hu, S., Du, G., Lin, W., Li, L., Bharill, S., Ness, R. A., Stecula, A., Offer, S. M., Diasio, R. B., Nies, A. T., Schwab, M., Cavaletti, G., Schlatter, E., Ciarimboli, G., Schellens, J. H., Isacoff, E. Y., Sali, A., Chen, T., Baker, S. D., Sparreboom, A. and Pabla, N. (2016). A phosphotyrosine switch regulates organic cation transporters. Nat Commun 7: 10880.
- Tanaka, K., Xu, W., Zhou, F. and You, G. (2004). Role of glycosylation in the organic anion transporter OAT1. J Biol Chem 279(15): 14961-14966.
- Zhang, Z., Yu, X., Wang, Z., Wu, P. and Huang, J. (2015). Anthracyclines potentiate anti-tumor immunity: A new opportunity for chemoimmunotherapy. Cancer Lett 369(2): 331-335.
- Zhou, F., Xu, W., Hong, M., Pan, Z., Sinko, P. J., Ma, J. and You, G. (2005). The role of N-linked glycosylation in protein folding, membrane targeting, and substrate binding of human organic anion transporter hOAT4. Mol Pharmacol 67(3): 868-876.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Brosseau, N., Andreev, E. and Ramotar, D. (2017). Uptake Assays to Monitor Anthracyclines Entry into Mammalian Cells. Bio-protocol 7(18): e2555. DOI: 10.21769/BioProtoc.2555.
Category
Cancer Biology > Cancer biochemistry > Genotoxicity
Cancer Biology > Genome instability & mutation > Cancer therapy
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.