- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Drosophila Fecal Sampling
Published: Vol 7, Iss 18, Sep 20, 2017 DOI: 10.21769/BioProtoc.2547 Views: 10320
Reviewed by: Jihyun KimAbhijit KaleAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2013
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Fecal sampling is a non-invasive method which raises the possibility to study the development and the changes in the microbial community throughout different time points of a fly population or throughout different treatments. This method allows precise manipulation to trigger the fly’s physiology by nutritional interventions, bacterial infections or other stressors.
As in most other animals, the intestinal microbiota is essential for a healthy fly-life. Because Drosophila only harbors a relative simple bacterial community with a small variety of round about 8 to 10 different species, it is rather easy to build up the microbial community and to investigate microbial changes after treatment.
Another positive effect using the fly’s feces is that bacteria that are not part of the intestinal microbiome, for example Wolbachia, can be excluded directly from the analysis because they are not excreted.
Using this method, the generated datasets may reflect a good paradigm to study microbiome associated diseases in a simple fly model or furthermore, to test drugs in a high-throughput approach.
Background
Until now, studies aiming to characterize the intestinal microbiome in the fruit fly Drosophila melanogaster used whole flies or dissected intestines as sources for bacterial DNA isolation. The main idea using feces sampling is to enable analyzing the dynamics of the intestinal microbiome in response to different treatments such as nutritional interventions or drug administration in the same cohort of flies. We were able to demonstrate that all general bacteria species, which are known to be important members of the Drosophila microbiome can be detected in fecal samples (Chandler et al., 2011; Wong et al., 2011; Matos and Leulier, 2014). Although whole fly samples are apparently the most convenient sources to generate microbiome data, several drawbacks are faced with this approach including contamination with surface bacteria or with intracellular bacteria such as Wolbachia spec (Saridaki and Bourtzis, 2010; Clark et al., 2005). Using feces as a source for microbiome data acquisition, these contaminations can be excluded almost completely, and, most importantly, the same cohorts of flies can be analyzed several times during the course of a complex experimental setup, thus unleashing the full potential of Drosophila genetics for microbiome studies.
Materials and Reagents
Note: For full name of the abbreviations in the text, please see Table S1 in Supplemental file.
- Filter tips, 0.5-10 µl, super slim, surface optimized (NERBE PLUS, catalog number: 07-613-8300 )
- Filter tips, 0-200 µl, super slim, surface optimized (NERBE PLUS, catalog number: 07-662-8300 )
- Filter tips, 0.5-10 µl, super slim, surface optimized (NERBE PLUS, catalog number: 07-695-8300 )
- Empty and clean Drosophila vials (NERBE PLUS, catalog number: 11-881-0052 )
- Cellulose plugs (NERBE PLUS, catalog number: 11-881-1010 )
- 0.22 μm sterilize filter filtropur (SARSTEDT, catalog number: 83.1826.001 )
- 1.5 ml reaction tubes (SARSTEDT, catalog number: 72.690.001 )
- 2 ml collecting tube
- Sterile spin swabs (Greiner Bio One International, catalog number: 420180 )
- X-tracta Tips (Biozym Scientific, catalog number: 615935 )
- Nitrogen (AIR LIQUIDE Deutschland, catalog number: I4001S10R2A001 )
- PowerSoil DNA isolation kit (NEW: Quiagen DNeasy PowerSoil Kit) (QIAGEN, catalog number: 12888 )
Note: Attention this kit was from MOBIO in the past, also called Power Soil Kit! - 70% ethanol diluted from 100% ethanol (Carl Roth, catalog number: 9065 )
- Proteinase K (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EO0491 )
- Phusion® Hot Start DNA polymerase (Thermo Fisher Scientific, catalog number: F540L )
- Crystal-Agarose (BIOLABPRODUCTS, catalog number: 16-005-805 )
- 100 bp DNA ladder (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15628019 )
- GeneRuler 50 bp DNA ladder (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SM0371 )
- qPCR Bio SYBR-Hi-ROX (NIPPON Genetics, catalog number: PB20.12-20 )
- Deoxynucleotide triphosphates (dNTPs) (Promega, catalog number: U1330 )
- Oligo dT 7 primer (custom made, order from Thermo Fisher Scientific)
- Oligonucleotides for target genes for use in qPCR (custom made, order from Thermo Fisher Scientific, use [20 µM])
- RT-PCR grade water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9935 )
- MinElute Gel Extraction Kit (QIAGEN, catalog number: 28604 )
- Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32851 )
- Agencourt AMPure XP (Beckman Coulter, catalog number: A63882 )
- Fly medium (see Recipes)
- Cornmeal (Mühle Schlingemann, catalog number: DAV-17080 )
- Brewer’s yeast (Leiber, catalog number: 17001636 )
- Glucose (Carl Roth, catalog number: 6780 )
- Agar-agar (Carl Roth, catalog number: 5210 )
- Sugar beat syrup (Kanne Brottrunk, catalog number: 2201 )
- Sugar molasses (Biohof Heidelicht, catalog number: 013aa )
- Cornmeal (Mühle Schlingemann, catalog number: DAV-17080 )
- Preservatives for fly medium:
- 10x phosphate-buffered saline (PBS) (see Recipes)
- 10x TBE (Tris-Borat-EDTA) buffer (see Recipes)
Equipment
- Eppendorf Research® plus, single-channel, variable, 0.1-2.5 µl, dark grey (Eppendorf, catalog number: 3120000011 )
- Eppendorf Research® plus, single-channel, variable, 0.5-10 µl, light grey (Eppendorf, catalog number: 3120000020 )
- Eppendorf Research® plus, single-channel, variable, 2-20 µl, yellow (Eppendorf, catalog number: 3120000038 )
- Eppendorf Research® plus, single-channel, variable, 100-1,000 µl, blue (Eppendorf, catalog number: 3120000062 )
- Autoclave
- Sterile bench (NuAire, model: NU-437-400E )
- Anesthetizing pistol (Blowgun) (GENESE Scientific, model: 54-104 )
- Dumont forceps #5 (Fine Science Tools, catalog number: 11252-20 )
- Scissors (Bioform, model: B37e )
- Vortexer (Scientific Industries, model: Vortex Genie 2 , catalog number: SI-0256)
- Vortex adapter (MO BIO, catalog number: 13000-V1 )
- Thermomixer (Eppendorf, model: Thermomixer Comfort )
- Centrifuge (Eppendorf, model: 5424 )
- Qubit 3.0 Fluorometer (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q33216 )
- Sensoquest labcycler (BIOLABPRODUCTS, catalog number: 11-011-101-096 )
- NanoDrop 3300 Fluorometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 3300, catalog number: ND-3300 )
- 25 °C incubator (AQUALYTIC, model: TC 140 G , catalog number: 438210)
- StepOne qPCR cycler (Thermo Fisher Scientific, catalog number: 4376357 )
- Gel DocTM XR+ Gel Documentation System (Bio-Rad Laboratories, model: Molecular Imager® Gel DocTM XR+, catalog number: 1708195 )
- Wide Mini-Sub® Cell GT Horizontal Electrophoresis System, 15 x 10 cm tray, with PowerPacTM Basic Power Supply (Bio-Rad Laboratories, catalog number: 1640301 )
- Agilent Bioanalyzer (Agilent Technologies, model: 2100 Bioanalyzer , catalog number: G2939BA)
- Water bath
- 1.5 L beaker
Software
- MOTHUR v1.23.1
- R statistics package v.2.13.1 (R_Development_Core_team, 2011)
Procedure
- Medium preparation
- Cook desired sterile fly medium (see Recipes) by autoclaving the medium at 120 °C for 20 min.
- Decontaminate empty and clean Drosophila vials and plugs under a safety cabinet via UV light for at least 30 min (Figure 1A).
- Let the medium cool down to 65 °C and add preservative agents under sterile conditions, if necessary.
- Pour the sterile fly medium into the decontaminated Drosophila vials under sterile conditions (Figure 1B).
- Let the medium dry under the safety cabinet and plug the vials (Figure 1C).
- It is possible to store the sterile medium at 4 °C in a sterile plastic box.

Figure 1. Preparation of sterile medium vials. A. Put empty (marked) vials and fresh plugs under the sterile bench and sterilize them using UV light for at least 30 min. B. Decant the fresh and sterile medium in the vials after decontamination under the bench and let the medium dry for about 45 to 60 min. C. Plug the vials. Vials can be kept at 4 °C in a sterile box.
- Cook desired sterile fly medium (see Recipes) by autoclaving the medium at 120 °C for 20 min.
- Feces collection
- Anesthetize the flies with nitrogen using the Blowgun (anesthetizing pistol). Transfer cohorts of 30 to 50 flies under sterile conditions to a sterile Drosophila vial and let them defecate for 24 h at 25 °C (Figure 2A).
- Then, transfer flies to a new vial under sterile conditions or discard them.
Note: Figures 2B and 2C show how vials and fecal spots will look like after 24 h.
Figure 2. Feces in the vial. Transfer 30 to 50 flies into a sterile prepared vial and let the flies defecate for about 24 h at 25 °C for standard wildtypes or crossings. B. After 24 h many fecal spots can be seen all over the vial (Figure 2C shows a magnification, red arrows mark only a few spots for better understanding). - Place sterile spin swabs, sterile 1x PBS (see Recipes), sterile scissors and the Drosophila vial containing the feces under the safety cabinet (Figure 3A).
- For better control, filtrate the needed amount of 1x PBS using a sterile filtropour filter (Figure 3B).
- Soak sterile spin swab with sterile 1x PBS (Figure 3C; for preparation step 5-8, see Video 1).
- Open vial and carefully collect the feces with the soaked spin swab.
Note: Don’t come in contact with the medium (Figures 3D and 3E).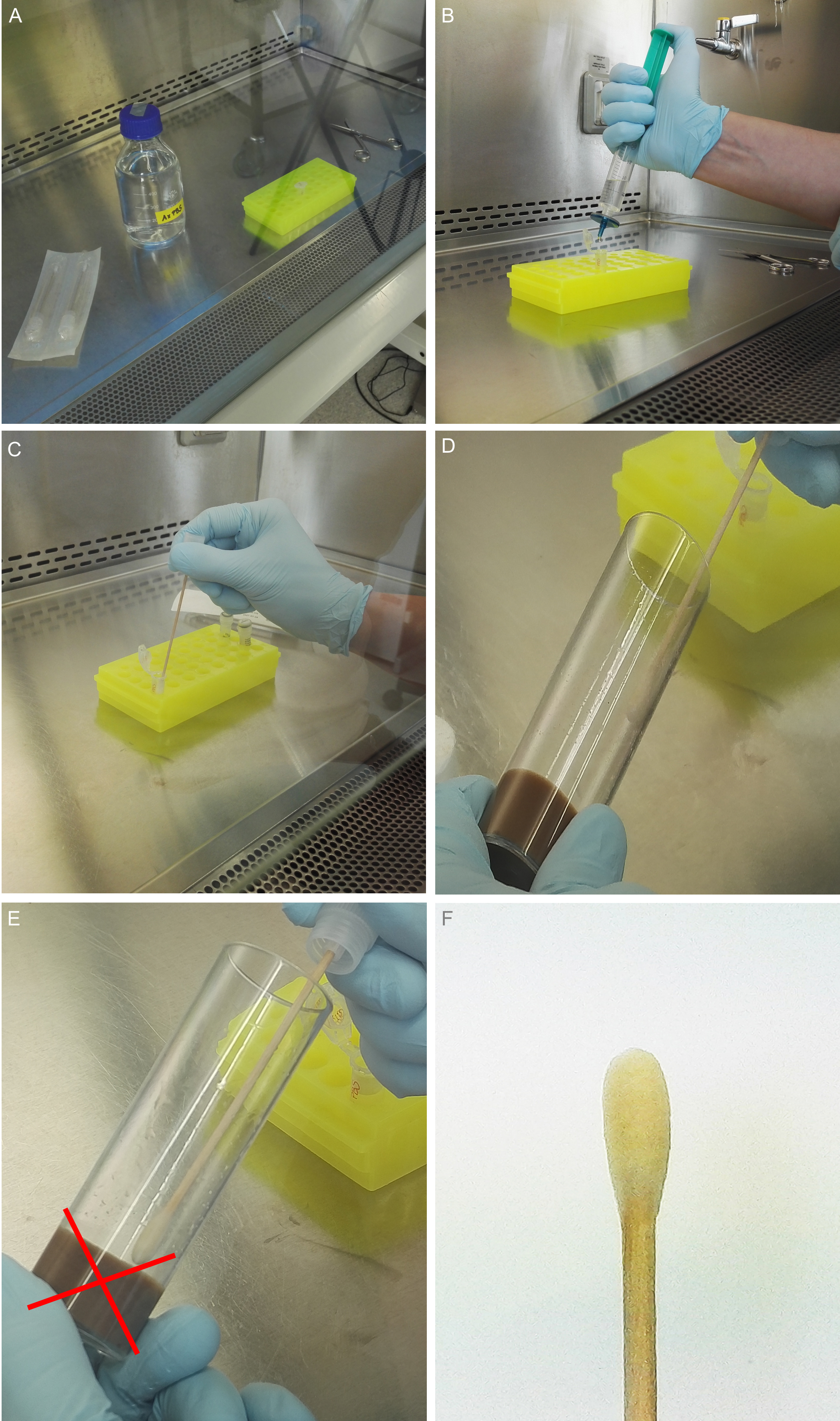
Figure 3. Feces collection. A. UV sterilization of all used materials, for example 1x PBS buffer, single packed swabs, sterile syringe, scissors and reaction tubes. B. Filter the needed amount of 1x PBS once again using a syringe filter. C. Soak the sterile swab with the fresh prepared and filtered 1x PBS. D. Start to collect/swab all fecal spots from the vial. ONLY from the walls, ignore spots on the plug AND take care NOT to touch the medium with the swab (E). F. Collected fecal spots from 40 flies after 24 h at 25 °C.
 Video 1. Feces collection
Video 1. Feces collection - Transfer the spin swab into a reaction tube and carefully cut off the stick with the sterile scissors (Figure 4C).
- Close the reaction tube.
- Prepare a negative control in the same way using an empty medium vial prepared at the same day as the used medium.
- Soak sterile spin swab with sterile 1x PBS (Figure 3C).
- Open vial and carefully collect the feces with the soaked spin swab (Figure 4A).
- After collection, the NTC (No Template Control) swab shows no feces (Figure 4B).
Note: Don’t come in contact with the medium (Figures 3D and 3E).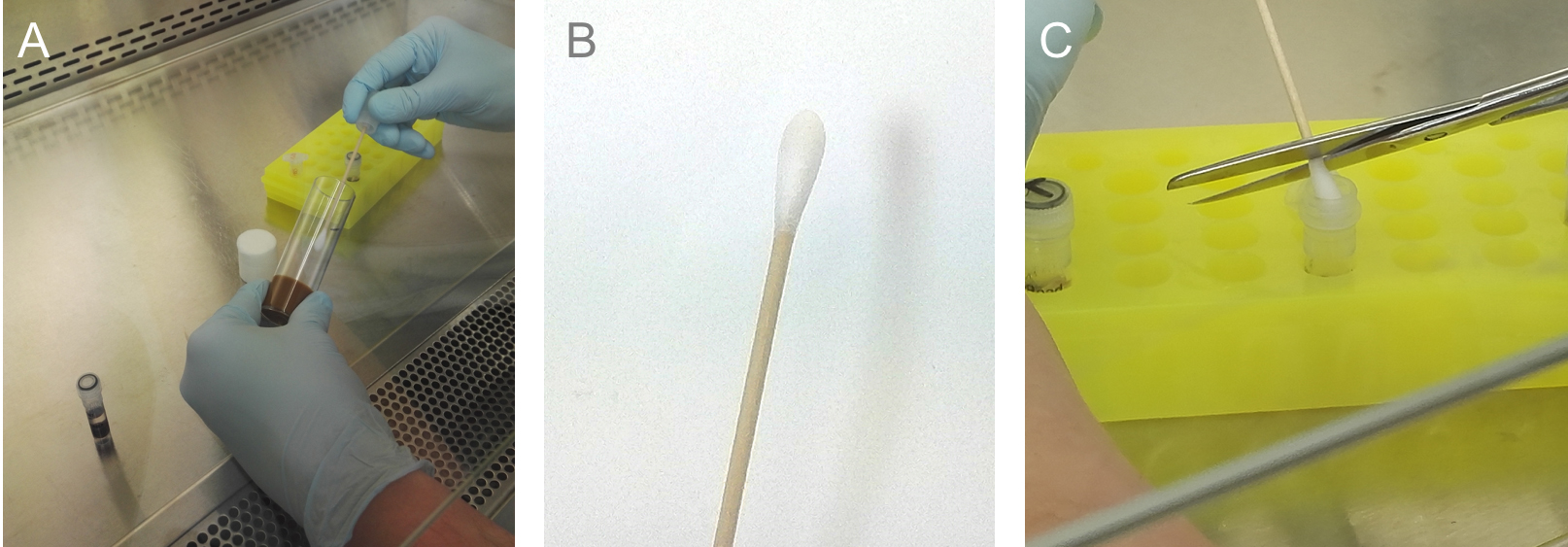
Figure 4. Negative control. A negative control (NTC), prepared at each time point of an isolation is necessary to check for a contamination of the vials and to have a baseline for later analysis. Start with the negative control always. A. Swab an empty medium vial (from the same charge as the treatment vials); B. Swab after ‘collection’; C. Transfer the swab into a Mobio Power Soil Tube.
- Anesthetize the flies with nitrogen using the Blowgun (anesthetizing pistol). Transfer cohorts of 30 to 50 flies under sterile conditions to a sterile Drosophila vial and let them defecate for 24 h at 25 °C (Figure 2A).
- gDNA isolation (manufacturer’s data with some simple modifications)
- Add 60 µl solution C1 (Power Soil Kit) and 20 µl Proteinase K to the reaction tube. Mix by inverting the reaction tube.
Note: If the solution is precipitated at room temperature, heat it up to 60 °C until it is homogenous again (Figures 5A-5C). - Incubate the sample for 2 h at 50 °C and 750 rpm in the thermomixer (Figure 5D).
- Vortex (vortex adapter) the samples at highest speed for 10 min (Figure 5E).

Figure 5. First steps of isolation using MOBIO Power Soil Kit. A. Add 60 µl of C1 (A, B) using sterile filter tips. C. Add 20 µl Proteinase K (STEP ADDED TO MANUFACTURERS PROTOCOL) and let the samples incubate in a thermos shaker (750 rpm) for 2 h at 50 °C (D, STEP ADDED TO MANUFACTURERS PROTOCOL) to induce Proteinase K activity. E. Transfer the vials into the MOBIO Vortex adapter and shake for 10 min at highest speed. F. The samples will look a lightly orange and may show a solid orange or red powder in the vial from the grinded silicate splints. This is absolutely normal. Go on by following the manufacturer’s protocol. - Centrifuge the sample at 10,000 x g for 30 sec at 20 °C.
Note: MAXIMUM 10,000 x g, tubes can break at higher rotations! - Pipette up to 400 µl of the supernatant in a sterile 2 ml collecting tube.
- Add 250 µl solution C2 (Power Soil) to the supernatant and vortex for 5 sec.
- Incubate the sample for 5 min at 4 °C and centrifuge at 10,000 x g for 60 sec at 20 °C.
- Pipette up to 550 µl of the supernatant in a sterile 2 ml collecting tube.
- Add 200 µl solution C3 (Power Soil) to the supernatant and vortex shortly.
- Incubate the sample for 5 min at 4 °C and centrifuge at 10,000 x g for 60 sec at 20 °C.
- Pipette not more than 600 µl of the supernatant in a sterile 2 ml collecting tube, add 1,200 µl of C4 (shake solution 4 well before using!) and vortex 5 sec.
- Because the maximum spin column capacity is 675 µl, pipette only 600 µl of the mixture of step C11 onto the column, centrifuge at 10,000 x g for 60 sec at 20 °C. Discard the flow through and repeat this step twice.
- Afterwards, wash the column by pipetting 500 µl of C5 onto the column.
- Centrifuge at 10,000 x g for 30 sec.
- Discard flow through and dry the column by centrifugation at 10,000 x g for 60 sec at 20 °C.
- Put the spin-filter (column) into a new sterile collecting tube.
- Pipette 30 µl of TE onto the middle of the column and incubate for 1 min.
- Centrifuge the column at 10,000 x g for 30 sec at 20 °C.
- Dispose of the spin-filter.
- gDNA can be stored at -20 °C.
- Add 60 µl solution C1 (Power Soil Kit) and 20 µl Proteinase K to the reaction tube. Mix by inverting the reaction tube.
- Further experiments
After gDNA extraction, use a Qubit 3.0 Fluorometer to measure the gDNA concentration. This gDNA can be used for further analysis by qPCR or MiSeq (454 sequencing described in Fink et al., 2013).
Notes:- You need at least 5 ng/µl for metagenomics.
- Check all fly strains for a Wolbachia infection before performing your experiments. In some cases, Wolbachia OTUs (Operating Taxonimic Units) will dominate in your 454 data sets of whole fly samples or gut samples. This is important if feces samples are compared with whole tissue samples.
- PCR amplifications
For PCR amplification we used 2.5 µl of gDNA as template in 25 µl reaction volume. Using 0.25 µl Phusion® Hot Start DNA polymerase, 0.5 µl of sense and antisense primer (10 mmol each); for PCR amplification, the Sensoquest labcycler was used. The general Wsp (Wolbachia specific) primers 81F (5’-TGGTCCAATAAGTGAAGAAAC-3’) and 691R (5’-AAAAATTAAACGCTACTCCA-3’) were used [10 mM] to amplify Wolbachia 16S RNA gene (Braig et al., 1994). To check the quality of the isolated DNA insect specific mitochondrial 12S ribosomal DNA primers were used, 12SAI (5’-AAACTAGGATTAGATACCCTATTAT-3’) and SBI (5’-AAGAGCGACGGGCGATGTGT-3’) (Simon et al., 1994). PCR conditions were: initial denaturation for 30 sec at 98 °C; 35 cycles of 9 sec at 98 °C, 30 sec at 55 °C, and 30 sec at 72 °C. The results are represented after gel electrophoresis in Figure 6 using 1.5 % Crystal-Agarose in 1x TBE (see Recipes).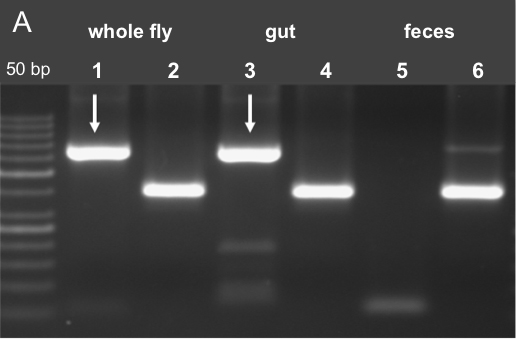
Figure 6. PCR analysis for Wolbachia contamination. A. PCR results using Wolbachia specific oligonucleotides (Wsp81F, Wsp691R) in lanes 1, 3 and 5. It is shown that fecal samples are free of Wolbachia contamination. To approve the success of DNA isolation gDNA specific 12S primers were used in lanes 2, 4 and 6 (12SAI and 12SBI) (Fink et al., unpublished data). - qPCR amplifications
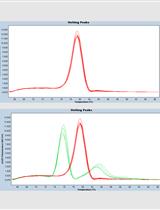
PCR was performed in 10 µl reactions containing 2 µl of gDNA template in a total reaction volume of 10 µl using 5 µl of the qPCR Bio SYBR-Hi-ROX. The general V2 primers were used as general detection primer or control primer [8 pmol] when comparing to other species specific amplification. PCR conditions using the StepOne Cycler were: Holding stage for 10 min at 95 °C; cycling stage 40 cycles of 10 sec at 95 °C, 30 sec at 60 °C annealing as time point for data collection. Melting curve stage: 10 sec at 95 °C, 5 sec at 60 °C, starting of data collection every 0.3 °C until the temperature of 95 °C was reached for 10 sec. - 454 procedures
- For PCR amplification of the 16sRNA gene a 311-nucleotide sequence flanking the hypervariable V1 and V2 region was used. For each sample, the 16S rRNA gene was amplified using the composite forward:
(5’-CTATGCGCCTTGCCAGCCCGCTCAGTCAGAGTTTGATCCTGGCTCAG-3’) and reverse (5’-CGTATCGCCTCCCTCGCGCCATCAGXXXXXXXXXXCATGCTGCCTCCCGTAGGAGT-3’) primers. These primers include the 454 Life Sciences Adaptor A (for reverse primer) and B (for forward primer)–(labeled with italics). In addition, barcodes of 10 bp (designated as XXXXXXXXXX) were added to the reverse primers. The underlined sequences represent the broadly conserved bacterial primers 27F and 338R. A two-base linker sequence (TC/CA) and four-base key (TCAG) were added (see Supplemental file). The amplification mix contained the Phusion® Hot Start DNA polymerase which was used following the standard protocol (initial denaturation for 30 sec at 98 °C; 35 cycles of 9 sec at 98 °C, 30 sec at 55 °C, and 30 sec at 72 °C; final extension for 10 min at 72 °C). See results after amplification in Figure 7. - All samples were done in duplicates. Duplicates were combined after PCR. PCR products were extracted with the QIAGEN MinElute Gel Extraction Kit and quantified with the Quant-iTTM dsDNA BR Assay Kit on a NanoDrop 3300 Fluorometer. Equimolar amounts of purified PCR product were pooled and further purified using Ampure Beads (Agencourt). A sample of each library was run on an Agilent Bioanalyzer prior to emulsion PCR (Rausch et al., 2011; SI) and sequencing as recommended by the manufacturer. Amplicon libraries were subsequently sequenced on a 454 GS-FLX using Titanium sequencing chemistry (Fink et al., 2013).
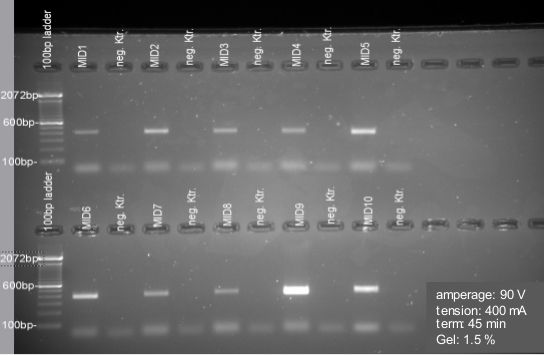
Figure 7. Gel electrophoresis for PCR quality check and Gel extraction. Samples show an amplicon size about 400 bp. Negative controls (neg. Ktr.) are included for each amplification. Samples are cut off by using the Gel DocTM XR+ X-tracta Tips. All represented negative controls are free of contamination here. If negative controls (NTC) show any contamination, re-run the samples in the PCR.
- For PCR amplification of the 16sRNA gene a 311-nucleotide sequence flanking the hypervariable V1 and V2 region was used. For each sample, the 16S rRNA gene was amplified using the composite forward:
- You need at least 5 ng/µl for metagenomics.
Data analysis
Note: For statistical analysis, you should provide at least three independent biological replicates for each treatment, it’s suggested that it is better to prepare five.
- qPCR-data analysis
We compared the Cycle threshold (Ct) from the PCR reaction, in which we used the primers specific to different bacterial species, to the Ct from the reactions with the general bacterial primers spanning the hypervariable regions V1 and V2 (V2 described above). By this relative comparison, we sought to control for the total amount of bacteria present in the sample. We averaged Cts between technical duplicates which resulted in a single Ct value per biological replicate using the following equation: ∆Ct = Ct (V2-primer set) - Ct Sample bacteria specific primer sets. The median ∆Ct of the three biological replicates was then used to transform this relative logarithmic measurement of bacterial abundance to a linear scale. We assumed doubling of the amount of PCR product with each cycle during the log-phase of the PCR reaction: x - fold = 2∆Ct (Fink et al., 2013). - 454-data analysis
- 454 reads were sorted into groups according to their multiplex identifier (MID) tags using MOTHUR v1.23.1 (Schloss, 2009). During the process, tags and primer sequences were removed. Only sequences matching the MIDs and the bacterial primers perfectly were kept. The resulting sequences were quality filtered according to the following conditions: Minimum average quality of 35 in each 50 base pair window, minimum length of 260 bp, homopolymers no longer than 8 bp. Filtered sequences were aligned to the SILVA reference database (Pruesse et al., 2007) using the MOTHUR implemented kmer (all the possible substrings of length k that are contained in a string) algorithm with standard settings. Sequences not aligning in the expected region were removed.
- Passing sequences were filtered for sequencing errors using the MOTHUR pre.cluster command. Sequences were then searched for chimeras using Uchime (Edgar et al., 2011) as implemented in MOTHUR with standard settings. Sequences identified as chimeras were discarded. Sequences were classified into bacterial taxa with the classify.seqs command in MOTHUR using the SILVA reference database and taxonomy. Results were plotted using the R statistics package v.2.13.1 (R_Development_Core_team, 2011). Clustering sequences into OTUs was performed using MOTHUR with the average neighbor algorithm. (Fink et al., 2013). An example stack plot is represented in Figure 8.
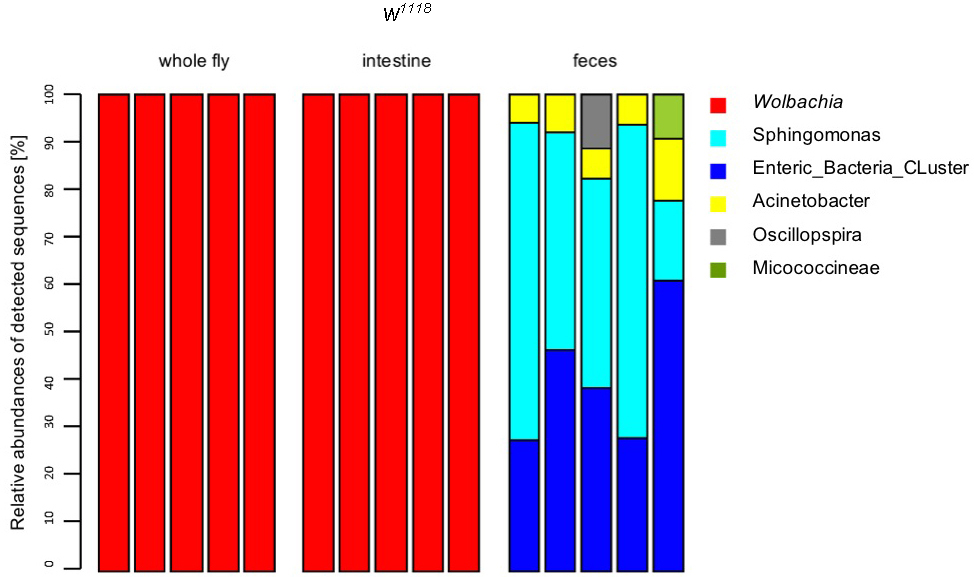
Figure 8. Stackplot of the relative abundance of OTUs detected after 454-sequencing. In the wildtype laboratory strain w1118 the Wolbachia infection/contamination could also be detected in the 454 data sets after sequencing (B, whole fly, gut samples). If there is a Wolbachia infection, most of the reads that are reflected by relative abundance (Y-axis) are reads from Wolbachia spec. This leads to the effect that it is not possible to find any other bacterial abundances in these samples. This will not affect fecal samples (Fink et al., unpublished data).
Notes
- Isolation is always reproducible. We could see in further analysis that there is no high variability between the samples of one treatment group. See Figure 8, feces samples: You nearly have the same abundance of all present bacteria. Sometimes, random sample dropouts may occur although the samples were extracted in the same run. Thus, we recommend to prepare always five samples of each treatment group.
- ALWAYS take care about a sterile environment, workspaces and tools. Any contamination could directly be seen in the further experiments.
- Take care NEVER touch the medium with the swab when doing feces collection (see Figure 3E).
Recipes
- Fly medium (1.5 L beaker)
62.5 g cornmeal
62.5 g brewer’s yeast
10 g agar-agar
20 g glucose
30 g molasses
20 g sugar beat syrup
Added in 1 L ddH2O
Cooking the medium for 10-15 min in a water bath at 100 °C
Autoclave for 15-20 min at 120 °C
Cool down the medium to 60 °C before adding the preservatives
30 ml Nipagin (10%)
10 ml propionic acid (10% in EtOH)
Prepare the vials under the clean bench. Add about 10 ml per vial - 10x phosphate-buffered saline (PBS)
80 g NaCl
2 g KCl
14.4 g Na2HPO4·2H2O
2.4 g KH2PO4
1 L ddH2O
Autoclave for 15-20 min at 120 °C
Adjust pH to 7.4
Note: Dilute the 10x stock buffer by 1 to 10 with ddH2O freshly before usage. - 10x TBE (Tris-Borat-EDTA) buffer
108 g Tris
55 g boric oxide
40 ml of 0.5 M Na2EDTA
1 L ddH2O
Autoclave for 15-20 min at 120 °C
Adjust pH to 8.0
Note: Dilute the 10x stock buffer by 1 to 10 with ddH2O freshly before usage.
Acknowledgments
We thank Britta Laubenstein, Christiane Sandberg and Katja Cloppenborg-Schmidt for their excellent technical support. This work was supported by the Deutsche Forschungsgemeinschaft as part of the CRC 1182 (Project C2) and the Cluster of Excellence Inflammation@Interfaces.
References
- Braig, H. R., Guzman, H., Tesh, R. B. and O’Neill, S. L. (1994). Replacement of the natural Wolbachia symbiont of Drosophila simulans with a mosquito counterpart. Nature 367(6462): 453-455.
- Chandler, J. A., Lang, J. M., Bhatnagar, S., Eisen, J. A. and Kopp, A. (2011). Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet 7(9): e1002272.
- Clark, M. E., Anderson C. L., Cande J. and Karr, T. L. (2005). Widespread prevalence of wolbachia in laboratory stocks and the implications for Drosophila research. Genetics 170(4): 1667-75.
- Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16): 2194-2200.
- Fink, C., Staubach, F., Kuenzel, S., Baines, J. F. and Roeder, T. (2013). Non-invasive analysis of microbiome dynamics in the fruit fly Drosophila melanogaster. Appl Environ Microbiol 79: 6984-6988.
- Huang, J. H., Jing, X. and Douglas, A. E. (2015). The multi-tasking gut epithelium of insects. Insect Biochem Mol Biol 67: 15-20.
- Matos, R. C. and Leulier, F. (2014). Lactobacilli-host mutualism: “learning on the fly”. Microb Cell Fact 13 Suppl 1: S6.
- Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J. and Glockner, F. O. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35(21): 7188-7196.
- Rausch, P., Rehmann, A., Keunzel, S., Haesler, R., Ott, S. J., Rosenstiel, P., Franke, A. and Baines, J. F. (2011). Colonic mucosa-associated microbiota is influenced by an interaction of Crohn’s disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A 108(47): 19030-5.
- Saridaki, A. and Bourtzis, K. (2010). Wolbachia: more than just a bug in insects genitals. Curr Opin Microbiol 13(1): 67-72.
- Schloss, P. D. (2009). A high-throughput DNA sequence aligner for microbial ecology studies. PLoS One 4(12): e8230.
- Simon, C., Frati, F., Beckenbach, A., Crespi, B., Liu, H. and Flook, P. (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87(6): 651-701.
- Wong, C. N., Ng, P. and Douglas, A. E. (2011). Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environ Microbiol 13(7): 1889-1900.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Fink, C., von Frieling, J., Knop, M. and Roeder, T. (2017). Drosophila Fecal Sampling. Bio-protocol 7(18): e2547. DOI: 10.21769/BioProtoc.2547.
Category
Microbiology > Microbe-host interactions > Bacterium
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.