- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Multicolor Stimulated Emission Depletion (STED) Microscopy to Generate High-resolution Images of Respiratory Syncytial Virus Particles and Infected Cells


(*contributed equally to this work) Published: Vol 7, Iss 17, Sep 5, 2017 DOI: 10.21769/BioProtoc.2543 Views: 10145
Reviewed by: Angela CoronaKristin ShinglerAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Human respiratory syncytial virus (RSV) infection in human lung epithelial A549 cells induces filopodia, cellular protrusions consisting of F-actin, that extend to neighboring uninfected cells (Mehedi et al., 2016). High-resolution imaging via stimulated emission depletion (STED) microscopy revealed filamentous RSV particles along these filopodia, suggesting that filopodia facilitate RSV cell-to-cell spread (Mehedi et al., 2016). In this protocol, we describe how to fix, permeabilize, immunostain, and mount RSV-infected A549 cells for STED imaging. We show that STED increases resolution compared to confocal microscopy, which can be further improved by image processing using deconvolution software.
Keywords: RSVBackground
RSV forms pleomorphic virus particles, with a predominance of long filaments about 100 nm in diameter and up to about 10 µm in length (Bachi and Howe, 1973; Mehedi et al., 2016). High-resolution light microscopy techniques are key to visualizing the interactions between RSV infected cells and virus particles. In a recent study, we used super-resolution fluorescence microscopy to study RSV cell-to-cell spread in human lung epithelial A549 cells.
STED microscopy is one of the super-resolution microscopy techniques that have been developed to circumvent the limitations imposed by the ~200 nm diffraction barrier of light (Hell and Wichmann, 1994; Westphal et al., 2008). STED is based on confocal fluorescence microscopy and employs a pair of lasers, namely a pulsed excitation source and a photon depletion source. The excitation pulse is focused on the sample and excites the fluorescent dye therein. The excitation laser is superimposed with a doughnut-shaped STED depletion laser that quenches excited dye molecules except for the doughnut hole at the very center of the excitation focus, so that emission occurs only from the narrow center. Narrowing the excitation focal point in this way allows for images to be taken at resolutions far below the diffraction limit, e.g., typically 30-80 nm. While STED imaging relies on efficient dye depletion, image resolution and intensity are limited by photobleaching inflicted upon the dye. To address these two contrasting, yet key issues that arise with STED imaging, optimal sample preparation, most notably dye selection and signal intensity optimization, are crucial. STED enabled us to state conclusively that RSV was attached to filopodia rather than merely in the vicinity, and to precisely enumerate viral particles. Here, we describe how samples were prepared for multicolor STED imaging including dye selection, fixation procedure, imaging parameters, and deconvolution. We show how STED and STED deconvolution can improve lateral resolution both qualitatively and quantitatively.
Materials and Reagents
- Aerosol resistant pipette tips
20 µl (Thermo Fisher Scientific, catalog number: 21-402-551 )
200 µl pipette tips (Thermo Fisher Scientific, catalog number: 02-682-255 )
1,000 µl pipette tips pipette tips (Thermo Fisher Scientific, catalog number: 21-402-582 ) - T225 cm2 flask with canted neck (Corning, Costar®, catalog number: 3001 )
- Microscope slides (super clean) (Scientific Device Laboratory, catalog number: 022 )
- Sterile 12 mm circle untreated cover glasses; thickness 0.13-0.17 mm (Carolina Biological Supply, catalog number: 633029 )
- 50 ml conical tube
- 24-well cell culture plate (Corning, Costar®, catalog number: 3524 )
- The cell line of interest (human respiratory epithelial A549 cells [ATCC, catalog number: CCL-185 ])
- Recombinant wild type RSV (A2 strain, GenBank KT992094) (virus stock with known virus titer, see Notes)
- TryLE Select cell dissociation reagent, stored at room temperature (Thermo Fisher Scientific, GibcoTM, catalog number: 12563 )
- Bovine serum albumin (BSA) standard (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 23210 )
- Anti-RSV F protein mouse monoclonal antibody (mAb) (Abcam, catalog number: ab43812 )
- Anti-beta-tubulin (9F3) rabbit mAb (Cell Signaling Technology, catalog number: 2128 )
- Goat anti-mouse Alexa Fluor 488 (AF488) (Thermo Fisher Scientific, catalog number: A11029 )
- Goat anti-rabbit IgG-Atto 647N (Sigma-Aldrich, catalog number: 40839 )
- Rhodamine phalloidin (CYTOSKELETON, catalog number: PHDR1 )
- ProLong Gold Antifade Mountant (Thermo Fisher Scientific, InvitrogenTM, catalog number: P36930 )
- Ultrapure methanol free formaldehyde prepared from paraformaldehyde (PFA) 16% solution, EM Grade (Polysciences, catalog number: 18814 )
- F-12 medium without additives, which is sold commercially as Ham’s F-12 nutrient mix (Thermo Fisher Scientific, GibcoTM, catalog number: 11765054 )
- Fetal bovine serum (FBS) (GE Healthcare, HyCloneTM, catalog number: SH30071.03 )
- L-Glutamine 200 mM (Thermo Fisher Scientific, GibcoTM, catalog number: 25030081 )
- Dulbecco’s phosphate buffer saline (DPBS) (Thermo Fisher Scientific, catalog number: 14190 )
- Triton X-100 (BioUltra, ~10% in H2O, Sigma-Aldrich, catalog number: 93443 )
- Trypan blue 0.4% solution (Lonza, catalog number: 17-942E )
- F-12 complete medium (see Recipes)
- 4% PFA (see Recipes)
- 0.05% Triton X-100 (see Recipes)
- 3% BSA (see Recipes)
Equipment
- Pipettes (Mettler-Toledo, RAININ, model: Pipet-Lite XLS )
- Humidified CO2 incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: FormaTM Steri-CultTM )
- Centrifuge (Beckman Coulter, model: Allegra 25R )
- Leica TCS SP8 STED 3X system (Leica Microsystems, model: Leica TCS SP8 STED 3X ) equipped with:
- A white light excitation laser
- 592 nm, 600 nm, 775 nm depletion lasers
- HC PL APO 100x/1.40 oil STED white objective
- Gated HyD hybrid detectors
- A white light excitation laser
- Hemocytometer (Marienfeld-Superior, catalog number: 0680030 )
- Dumont NOC tweezer (Electron Microscopy Sciences, catalog number: 0103-NOC-PO-1 )
Software
- Images were acquired using LAS X software (version 3.1.1.15751) (Leica Microsystems)
- Images were deconvolved using Huygens deconvolution software (Huygens Essentials version 16.10.1.p3, Scientific Volume Imaging BV, Hilversum, The Netherlands)
- PRISM software version 7
Procedure
- Virus infection
- Maintain A549 cells in T225 flasks, following the ATCC’s culturing recommendations, with the following modifications:
- Use F-12 complete medium to maintain the line (see Recipes); use TrypLE Select cell dissociation reagent; use FBS to inactivate the TrypLE Select.
- To seed A549 cells onto coverslips, remove the F12 complete cell culture medium from a T225 flask containing a monolayer of about 80% confluent A459 cells. Gently rinse the monolayer with 10 ml pre-warmed (37 °C) 1x DPBS, and remove the DPBS.
- To dissociate cells, add 5 ml TryLE Select cell dissociation reagent, and incubate for 5 min at 37 °C in a standard humidified CO2 incubator. Inactivate TrypLE Select by adding 10 ml of cold FBS. Detach and dissociate A549 cells by gentle pipetting, and transfer the single-cell suspension to a 50 ml conical tube.
- Pellet the cells by centrifugation at 300 x g for 5 min at 4 °C in a tabletop centrifuge. Resuspend the cell pellet in 10 ml of pre-warmed (37 °C) F-12 complete medium. Dilute 10 µl of the resuspended A549 cells 1:10 in DPBS with 0.04% trypan blue (80 µl DPBS,10 µl trypan blue [0.4%],10 µl aliquot of A549 cells, resuspended in complete F-12 medium) and determine the concentration of the resuspended cells per ml using a hemocytometer as recommended by the manufacturer.
- Dilute the resuspended A549 cells from the 50 ml conical tube with F-12 complete medium to 3 x 104 cells per ml, and seed cells by adding 1 ml of F-12 complete medium with 3 x 104 cells to each well of a 24-well plate containing a coverslip.
- Incubate the cells overnight in a cell culture incubator at 37 °C, 5% CO2.
- Use F-12 complete medium to maintain the line (see Recipes); use TrypLE Select cell dissociation reagent; use FBS to inactivate the TrypLE Select.
- To infect A549 cells, replace the medium with 150 µl F-12 medium without additives, containing sucrose-purified RSV (Collins et al., 1995 and Le Nouen et al., 2009; see Notes) at a multiplicity of infection (MOI) of 1 plaque forming unit (PFU) per cell (see Notes).
- Incubate A549 cells with virus inoculum for 1 h at 37 °C in a CO2 incubator.
- Remove virus inoculum and wash infected cells 2 x with 1 ml F-12 medium without additives.
- Incubate infected cells in 1 ml of F-12 medium with 2% FBS and 1% L-glutamine for 24 h at 37 °C in a CO2 incubator.
- Maintain A549 cells in T225 flasks, following the ATCC’s culturing recommendations, with the following modifications:
- Slide preparation
- Remove cell culture medium and wash monolayers 3 x with 1 ml DPBS.
- To fix infected cells, incubate cells with 1 ml of a freshly prepared 1:4 dilution of PFA (4% final concentration) in DPBS for 30 min at room temperature.
- Remove PFA solution and wash cells 3 x each with 1 ml DPBS.
- To permeabilize fixed cells, incubate with 1 ml 0.05% Triton X-100 in DPBS for 10 min at room temperature.
- Remove Triton X-100 solution and wash cells 3 x each with 1 ml DPBS.
- To block unspecific protein binding, incubate coverslip with 1 ml 3% BSA in PBS for 3 h at 4 °C.
- Wash cells 1 x with 1 ml DPBS.
- Incubate cells overnight at 4 °C with primary antibody mix: mouse anti-RSV F mAb (1:500) and rabbit anti-tubulin mAb (1:100) in PBS with 0.1% BSA.
- Wash cells 3 x each with 1 ml DPBS.
- Incubate cells with secondary antibody mix: goat anti-mouse AF488 (1:200) and goat anti-rabbit Atto 647N (1:100) in DPBS with 0.1% BSA for 3 h at 4 °C in the dark.
- Wash cells 3 x each with 1 ml DPBS.
- For actin cytoskeleton staining, incubate cells with rhodamine phalloidin (1:500) in DPBS for 30 min at 4 °C in the dark.
- Wash cells 2 x each with 1 ml DPBS.
- Wash cells 2 x each with 1 ml deionized H2O.
- To mount the coverslip on a glass slide, first place 10 µl of ProLong Gold Antifade mounting medium onto a glass slide. Pick up the coverslip with Dumont #NO forceps and lower the cover slip cell-side down onto the mounting medium, taking care to avoid trapping air bubbles.
- Dry slide overnight in the dark.
- Remove cell culture medium and wash monolayers 3 x with 1 ml DPBS.
- Imaging procedure
- In Figure 1, all images were collected in a single focal plane with a 30 nm pixel size using a bidirectional scan speed of 600 Hz.
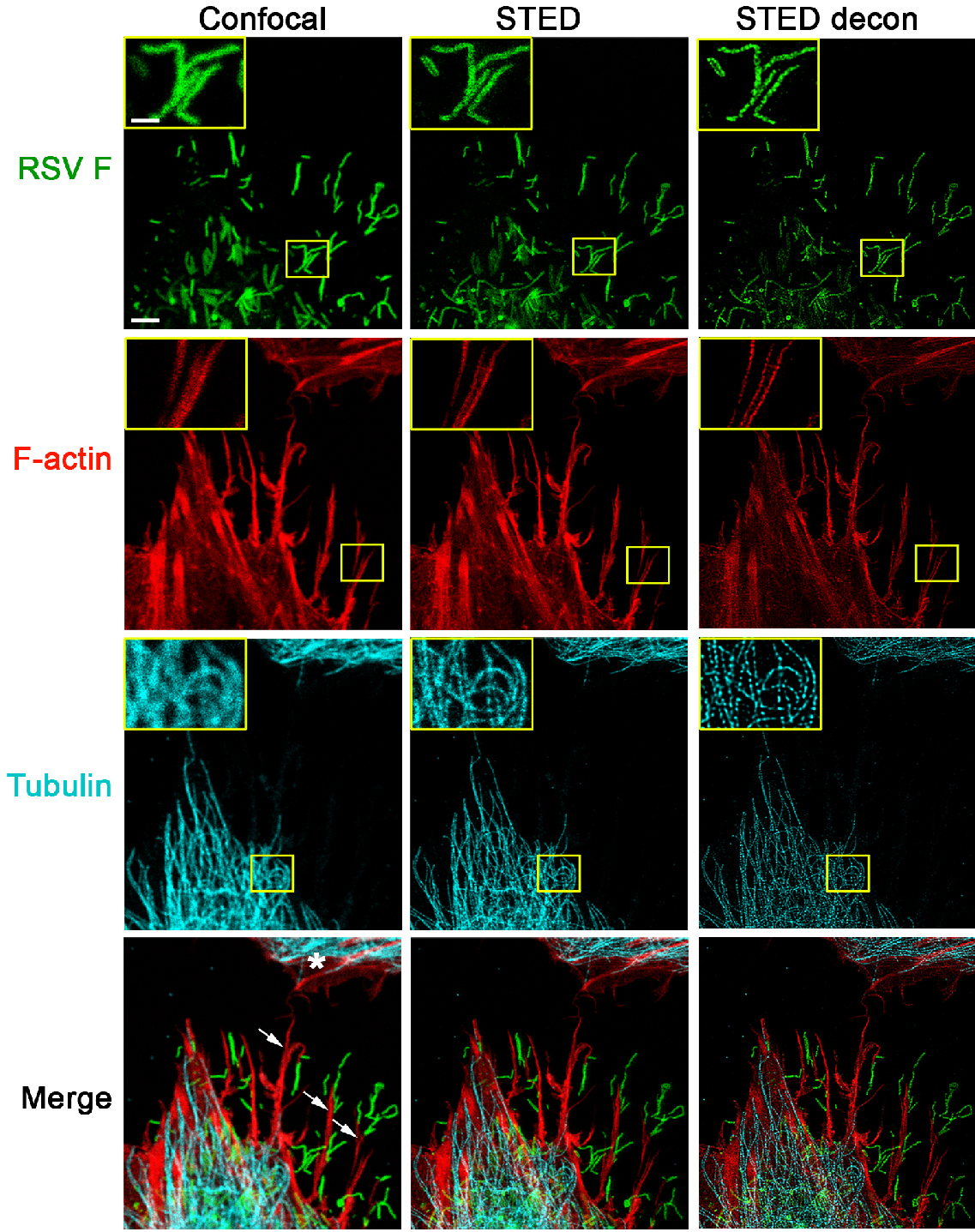
Figure 1. STED imaging of RSV viral particles along filopodia. A549 cells were infected with RSV (MOI = 1 PFU/cell) for 24 h. Cells were fixed, permeabilized, and immunostained with antibodies for RSV F (green); the cellular tubulin network was visualized by staining for beta tubulin (cyan). These cells were further stained with rhodamine phalloidin to detect F-actin (red). All images were collected in a single focal plane as described in the imaging procedure. For confocal image acquisition (left panel), excitation wavelengths for the detection of RSV F (AF488), F-actin (rhodamine phalloidin), and tubulin (Atto 647N) were 488 nm, 561 nm, and 647 nm, respectively. The subsequent STED acquisition (middle panel) was done in the following order: first, the Atto 647N conjugate used for tubulin immunostaining was excited with 647 nm and depleted with 775 nm; then, to visualize F-actin, rhodamine phalloidin was excited with 561 nm and depleted with 660 nm; and finally, the AF488 conjugate used for RSV F specific immunostaining was excited with 488 nm and depleted with 592 nm. Filopodia (indicated by arrows on the bottom left image) appear to shuttle RSV particles towards a neighboring cell (marked with a * in the bottom left image). An increase in resolution of all channels is apparent with STED imaging, which can be further improved by deconvolution using Huygens software (STED decon, right panel). Scale bar = 3 µm, inset scale bar = 1 µm. - Gated HyD detectors were used to collect an emission bandwidth of approximately 40 nm. All fluorophores were excited with a pulsed white light laser tuned to the appropriate wavelength.
- For confocal channel acquisition, AF488 was excited with 488 nm; rhodamine phalloidin was excited with 561 nm; and Atto 647N was excited with 647 nm. All HyD detector gating was set to 0.3-6 nsec. High intensity signals enabled the usage of low laser power settings, and single color controls were used to confirm the absence of background fluorescence. Of note, the laser power for confocal acquisition was set 3-5 fold lower than for STED acquisition. Photobleaching during confocal acquisition was negligible.
- Photon depletion occurs when some overlap exists between the STED laser wavelength and the emission spectrum of the fluorophore. Thus, for STED channel acquisition (subsequent to confocal acquisition), AF488 was excited with 488 nm and photon depleted with 592 nm with 1.2-6 nsec HyD gating; rhodamine phalloidin was excited with 561 nm and depleted with 660 nm with 1-6 nsec HyD gating; and Atto 647N was excited with 647 nm and depleted with 775 nm with 0.6-6 nsec HyD gating.
- Caution needs to be taken to limit exposure of fluorophores to a depletion laser if they can absorb energy at that specific wavelength (i.e., fluorophores that have excitation spectrums encompassing the wavelengths of the depletion lasers, 592 nm, 660 nm, or 775 nm). This is because the depletion laser output at the imaging plane is ~500-1,000 times greater than the excitation source, which will rapidly result in photobleaching. For example, Atto 647N will be photobleached by the 660 nm depletion laser, Alexa 594 will be photobleached by the 592 nm depletion laser, etc. Therefore, to avoid photobleaching, the collection order of the STED channels is of utmost importance, with longer wavelength fluorophores collected first and the shorter wavelengths collected last. In this experiment, the collection order of the STED channels was Atto 647N, followed by rhodamine phalloidin, and then AF488.
- Using single color controls, we confirmed that only the intended fluorophore was excited per each channel, and that neither cellular autofluorescence nor non-specific binding of the primary or secondary antibodies were detected.
- While confocal images were collected with a pinhole set to 1 Airy Unit (AU), this was reduced to 0.7 AU for STED imaging to reduce optical sectioning and increase the signal-to-noise ratio.
- Additionally, due to the strong depletion power, excitation powers were increased approximately 3-5 fold compared to confocal to compensate for signal loss. A frame accumulation of 2 was also used to further amplify the signal.
- In Figure 1, all images were collected in a single focal plane with a 30 nm pixel size using a bidirectional scan speed of 600 Hz.
- Image processing
- STED images were deconvolved using Huygens deconvolution software (Huygens Essentials v.17.040.p5, SVI BV, The Netherlands) to reverse the optical distortion created during image acquisition (Figure 2). Within this software package, we used the Deconvolution Wizard with automatic background subtraction and microscopic parameters recognition with a continued maximum likelihood estimate (CMLE) iterative algorithm. Processing parameters included microscopic and deconvolution parameters, which are important for proper point spread function (PSF) calculation necessary for successful deconvolution. Microscopic parameters were verified and corrected if necessary to avoid processing artifacts. The deconvolution parameters were adjusted in this Huygens deconvolution package as described below. This Huygens Essentials package was the only STED deconvolution package available at that time. However, there now are a number of software packages or free plug-ins available for classical confocal, multiphoton, or wide-field deconvolution applications.
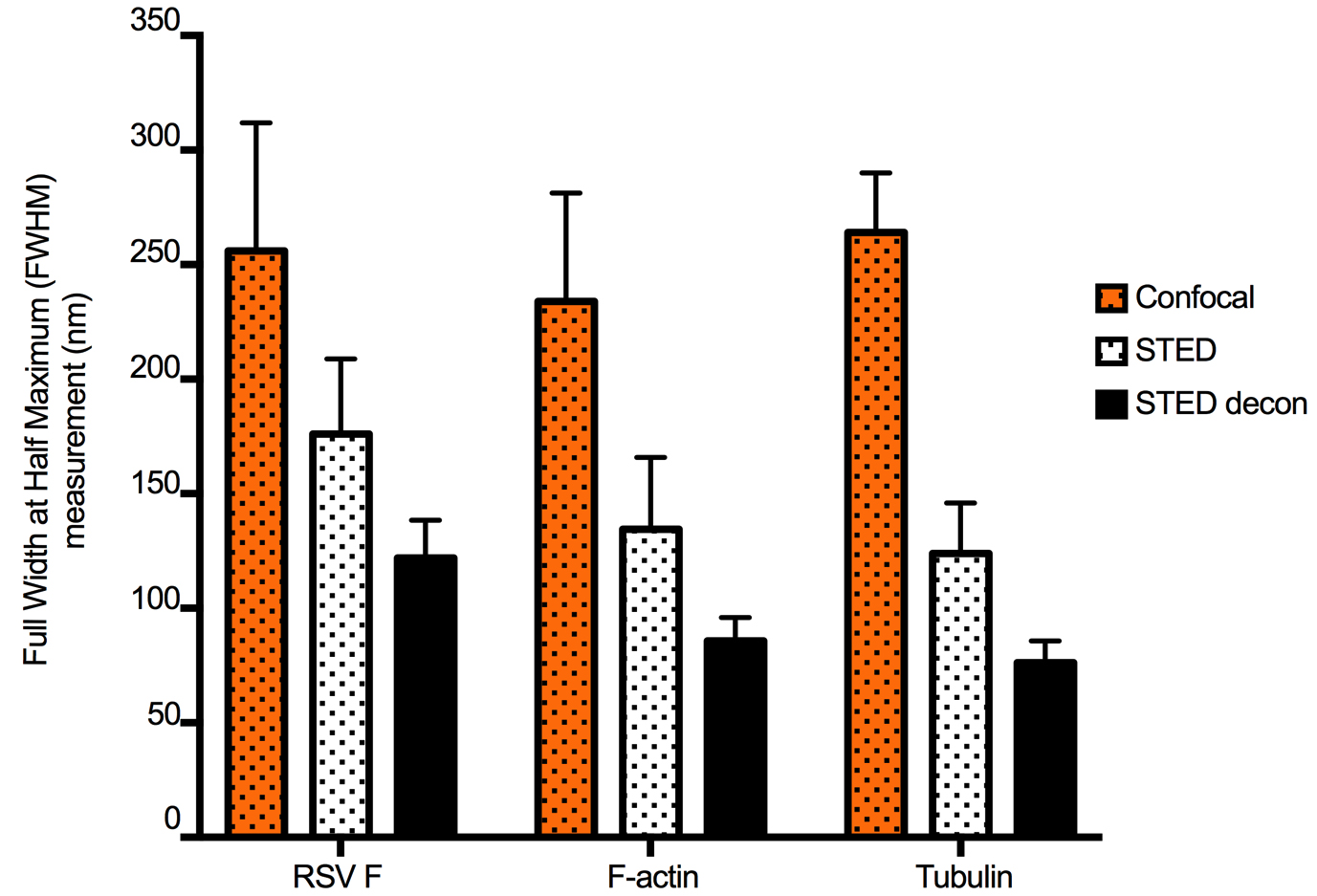
Figure 2. STED and deconvolution improve lateral resolution. To quantify the improvement in image resolution shown in Figure 1, we took the Gaussian profiles (bell curves) of signal intensity for a number of representative narrow structures and measured the width of the curve at the intensity level that is half of the maximum, which provided ‘full-width at half-maximum’ (FWHM) values. Smaller values of FWHM indicate improved resolution. This was done for five structures from each channel (green: virus filaments (RSV F), red: actin filaments, and cyan: tubulin filaments). The same structures were measured in confocal, STED, and STED deconvolved (STED decon) images. For each stain, STED significantly improved resolution compared to confocal microscopy, and resolution was further improved by deconvolution. Error bars represent standard deviation. - STED resolution was improved in Huygens Essentials by using settings for background subtraction, for the number of iterations, and for the desired signal-to-noise ratios that were determined empirically after several rounds of iterations. For RSV F, these values were 0.0316 for background subtraction, 54 for the number of iterations, and 18 for the signal-to-noise ratio. For F-actin, these values were 0.0695, 46, and 19, respectively. For tubulin, these values were 0.0835, 49, and 20, respectively. Deconvolved images were thoroughly compared with the original raw image to avoid artifacts such as striping, ringing, or discontinuous staining.
- Additional deconvolution parameters were set in Huygens Essentials to account for the expected amount by which the fluorescence is suppressed by the STED beam (STED saturation factor) and the expected fraction of fluorescent molecules that is photoresistant to the depletion beam (STED immunity fraction). The STED saturation factor is the absolute intensity of the STED laser divided by the saturation intensity. For the maximum intensity STED laser, the saturation factor is 30, and it is scaled based on the STED laser intensity. The STED immunity fraction is the fraction of the fluorophores that has not been depleted by the STED laser. It is described as an additional confocal PSF component that is added to the pure STED PSF and is estimated in percentage of the maximum saturation. It has only a minor influence on the quality of deconvolved images, and typical values are around 10%. For RSV F, these values were 30 for the STED saturation factor, and 14 for the STED immunity fraction. For F-actin, these values were 27 and 10, respectively. For tubulin, these values were 5 and 11, respectively.
- STED images were deconvolved using Huygens deconvolution software (Huygens Essentials v.17.040.p5, SVI BV, The Netherlands) to reverse the optical distortion created during image acquisition (Figure 2). Within this software package, we used the Deconvolution Wizard with automatic background subtraction and microscopic parameters recognition with a continued maximum likelihood estimate (CMLE) iterative algorithm. Processing parameters included microscopic and deconvolution parameters, which are important for proper point spread function (PSF) calculation necessary for successful deconvolution. Microscopic parameters were verified and corrected if necessary to avoid processing artifacts. The deconvolution parameters were adjusted in this Huygens deconvolution package as described below. This Huygens Essentials package was the only STED deconvolution package available at that time. However, there now are a number of software packages or free plug-ins available for classical confocal, multiphoton, or wide-field deconvolution applications.
Notes
- To avoid disturbing the cell monolayer while pipetting, tilt the plate slightly (at an angle less than 45°) and direct the pipette tip towards the side wall of each well during dispensing.
- Pipette slowly to preserve fragile cellular structures and viral filaments.
- To prevent possible interfering effects on cell biology, do not include antibiotic and anti-fungal agents when culturing A549 cells
- It is not essential that sucrose purified recombinant RSV be used for these studies.
- Prior to these studies, the titer of the RSV stock should be determined by immunoplaque assay on 24-well plates of Vero 76 cells (ATCC CRL-1587). In short, prepare serial tenfold dilutions of the RSV stock of interest in cell culture medium. Discard the growth medium from 24-well plates of subconfluent Vero cell monolayers, and transfer 100 µl per well of the serial RSV dilutions to duplicate wells of 24-well plates. To allow for virus adsorption, incubate for 2 h in a cell culture incubator; gently rocking the plates every 20 min to prevent the monolayers from drying out. After adsorption, overlay the monolayers with 1 ml of 0.8% methyl cellulose overlay (prepared using cell culture medium) per well, and incubate the cultures for 5-6 days. Then discard the methyl cellulose overlay by inverting the plates, and fix with ice-cold 80% methanol. Visualize RSV plaques by immunostaining using an RSV specific primary antibody preparation (for example a commercially available mouse monoclonal antibody to RSV), followed by a species-specific secondary antibody (for example, a peroxidase-conjugated goat anti-mouse IgG(H+L), KPL #074-18064), and a detection system of choice (for example, CN Peroxidase substrate, KPL #50-73-02, and peroxidase substrate solution B, KPL #50-65-02). Select wells of a dilution that yielded well-separated plaques, count virus plaques per well, and calculate the titer of the virus stock (plaque forming units per ml) by multiplying the average number of plaques by the dilution factor.
Data analysis
Images were deconvolved and FWHM measurements were made using Huygens deconvolution software. Standard deviation was calculated using PRISM software version 7. STED microscopic observation of filopodia-driven RSV cell-to-cell spread has previously been described in detail in (Mehedi et al., 2016). Here, we include an additional channel to visualize the tubulin network in the RSV infected cells.
Recipes
- F-12 complete medium
500 ml F-12 nutrient mix
10% FBS
1% L-glutamine - 4% PFA
30 ml 1x DPBS
10 ml 16% PFA - 0.05% Triton X-100
50 ml 1x DPBS
250 µl Triton X-100 - 3% BSA
50 ml 1x DPBS
1,500 µl BSA (1 mg/ml)
Acknowledgments
This study was supported by the Intramural Research Program of NIAID, NIH. All authors have declared that no competing interest exists. This protocol was adopted from previously published work (Mehedi et al., 2016).
References
- Bachi, T. and Howe, C. (1973). Morphogenesis and ultrastructure of respiratory syncytial virus. J Virol 12(5): 1173-1180.
- Collins, P. L., Hill, M. G., Camargo, E., Grosfeld, H., Chanock, R. M. and Murphy, B. R. (1995). Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5’ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc Natl Acad Sci U S A 92(25): 11563-11567.
- Hell, S. W. and Wichmann, J. (1994). Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 19(11): 780-782.
- Le Nouen, C., Munir, S., Losq, S., Winter, C. C., McCarty, T., Stephany, D. A., Holmes, K. L., Bukreyev, A., Rabin, R. L., Collins, P. L. and Buchholz, U. J. (2009). Infection and maturation of monocyte-derived human dendritic cells by human respiratory syncytial virus, human metapneumovirus, and human parainfluenza virus type 3. Virology 385(1): 169-182.
- Mehedi, M., McCarty, T., Martin, S. E., Le Nouen, C., Buehler, E., Chen, Y. C., Smelkinson, M., Ganesan, S., Fischer, E. R., Brock, L. G., Liang, B., Munir, S., Collins, P. L. and Buchholz, U. J. (2016). Actin-related protein 2 (ARP2) and virus-induced filopodia facilitate human respiratory syncytial virus spread. PLoS Pathog 12(12): e1006062.
- Westphal, V., Rizzoli, S. O., Lauterbach, M. A., Kamin, D., Jahn, R. and Hell, S. W. (2008). Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science 320(5873): 246-249.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mehedi, M., Smelkinson, M., Kabat, J., Ganesan, S., Collins, P. L. and Buchholz, U. J. (2017). Multicolor Stimulated Emission Depletion (STED) Microscopy to Generate High-resolution Images of Respiratory Syncytial Virus Particles and Infected Cells. Bio-protocol 7(17): e2543. DOI: 10.21769/BioProtoc.2543.
Category
Microbiology > Microbe-host interactions > Virus
Microbiology > Microbe-host interactions > In vivo model > Mammal
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.