- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation and Separation of Epithelial CD34+ Cancer Stem Cells from Tgfbr2-deficient Squamous Cell Carcinoma


Published: Vol 7, Iss 17, Sep 5, 2017 DOI: 10.21769/BioProtoc.2524 Views: 9929
Reviewed by: Nicoletta CordaniShweta GargAnonymous reviewer(s)
Original research article
The authors used this protocol in:

13-Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Most epithelial tumors have been shown to contain cancer stem cells that are potentially the driving force in tumor progression and metastasis (Kreso and Dick, 2014; Nassar and Blanpain, 2016). To study these cells in depth, cell isolation strategies relying on cell surface markers or fluorescent reporters are essential, and the isolation strategies must preserve their viability. The ability to isolate different populations of cells from the bulk of the tumor will continue to deepen our understanding of the biology of cancer stem cells. Here, we report the strategy combining mechanical tumor dissociation, enzymatic treatment and flow cytometry to isolate a pure population of epithelial cancer stem cells from their native microenvironment. This technique can be useful to further functionally profile the cancer stem cells (RNA sequencing and epigenetic analysis), grow them in culture or use them directly in transplantation assays.
Keywords: Cancer stem cellsBackground
Tumor recurrence and metastasis is the leading cause of most deaths related to cancer. Malignant tumors may be initiated and maintained by a stem cell population (Nassar and Blanpain, 2016; Bonnet and Dick, 1997), and these cells represent an important therapeutic target to prevent relapse (Baumann et al., 2008). Studies suggest that squamous cell carcinomas are maintained by a subpopulation of tumor cancer stem cells that are resistant to therapy and can initiate tumor recurrence by undergoing self-renewal and differentiation, like normal stem cells, giving rise to proliferating progenitor cells that differentiate and form the bulk of the tumor (Locke et al., 2005; Prince et al., 2007; Malanchi et al., 2008; de Sousa e Melo et al., 2017). In this scenario, tumor cell fate and behavior are determined by the specific combination of changes in genes or their expression that have occurred during tumor development (Wang, 2010). Alternatively, progenitor cells can also acquire mutations that give them the potential to self-renew or can acquire some plasticity that give them cancer stem cell properties (Shimokawa et al., 2017). Regardless of the origin of cancer stem cells, efficient techniques to isolate these cells while maintaining their viability is essential. We have extensively characterized cancer stem cells from anorectal transition zone squamous cell carcinoma which arise spontaneously in the absence of epithelial TGFβ signaling (Keratin14Cre; Tgfbr2flox/flox mice) (Guasch et al., 2007; McCauley and Guasch, 2013; McCauley et al., 2017 ). In this protocol, we describe a method to isolate cancer stem cells from these anorectal transition zone squamous cell carcinomas.
Materials and Reagents
- Tissue culture dishes, 100 x 20 mm (Corning, Falcon®, catalog number: 353003 ) and 60 x 15 mm (Corning, Falcon®, catalog number: 353002 )
- Sterile disposable scalpel, #21 (Sklar)
- 50 ml conical tubes (BD, Falcon)
- Pipettes (25 ml, 10 ml and 5 ml serological and 1,000 µl, 200 µl, 20 µl and 2 µl pipette tips)
- Sterile nylon cell strainers, 70 μm (Fisher Scientific, catalog number: 22-363-548 ) and 40 µm (Fisher Scientific, catalog number: 22-363-547 )
- 5 ml polystyrene round-bottom tubes with cell strainer caps, 12 x 75 mm style (Corning, Falcon®, catalog number: 352235 )
- Sterile screw cap tubes and caps with ‘O’ rings, 1.5 ml (Corning, Axygen®, catalog number: SCT-150-C-S )
- Nalgene sterile disposable vacuum filters (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 167-0045 )
- Healthy and Tumor-bearing mice
- For healthy control mice, we used any mouse in the colony that did not express a transgene
- We used Keratin14Cre; Tgfbr2flox/flox; R26R-eYFPSTOP-flox-STOP mice, which spontaneously develop anorectal squamous cell carcinoma (Guasch et al., 2007; McCauley and Guasch, 2013; McCauley et al., 2017) in the development of this protocol. All three alleles are available from Jackson Labs (THE JACKSON LABORATORY, catalog number: 018964 , 012603 , and 006148 , respectively)
- 1x Hanks’ balanced salt solution (HBSS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14170112 )
- Deoxyribonuclease (DNAse) I, from bovine pancreas, 10 mg/ml (Sigma-Aldrich, catalog number: D4263-5VL )
- 1x phosphate-buffered saline, sterile (1x PBS) (made in-house)
- Trypsin-EDTA, 0.25% (Thermo Fisher Scientific, GibcoTM, catalog number: 25200056 ) pre-warmed to 37 °C
- 7-AAD, 0.05 mg/ml (BD, BD Biosciences, catalog number: 559925 )
- Antibodies
- PE-Cy7 rat-anti-mouse CD11b, clone M1/70, 1/200 dilution (BD, BD Biosciences, catalog number: 561098 , RRID:AB_ 2033994)
- PE-Cy7 rat-anti-mouse CD31, clone 390, 1/100 dilution (BD, BD Biosciences, catalog number: 561410 , RRID:AB_10612003)
- PE-Cy7 rat-anti-mouse CD45, clone 30-F11, 1/200 dilution (Thermo Fisher Scientific, eBioscienceTM, catalog number: 25-0451-82 , RRID:AB_469625)
- PE rat-anti-human CD49f, 1/50 dilution (BD, BD Biosciences, catalog number: 555736 , RRID AB_396079)
- Pacific Blue anti-mouse/rat CD29, clone HMb1-1, 1/100 dilution (BioLegend, catalog number: 102224 , RRID:AB_2128079)
- Biotin anti-mouse CD34, clone RAM34, 1/50 dilution (Thermo Fisher Scientific, eBioscienceTM, catalog number: 13-0341-85 , RRID:AB_ 466426)
- APC Streptavidin, 1/200 dilution (BD, BD Bioscience, catalog number: 554067 , RRID:AB_10050396)
- β-Mercaptoethanol (Sigma-Aldrich, catalog number: M3148 )
- QIAGEN RNeasy Micro kit (QIAGEN, catalog number: 74004 )
- Matrigel Matrix Basement Membrane, 5 ml vial (Corning, catalog number: 356234 )
- Sterile distilled, deionized water
- Collagenase (Sigma-Aldrich, catalog number: C2674-1G )
- 20% collagenase from Clostridium histolyticum (see Recipes)
- Chelated fetal bovine serum (FBS) (see Recipes) (Nowak and Fuchs, 2009)
- Chelex, 100 resin, sodium, 200-400 dry mesh, 75-150 µm wet bead (Bio-Rad Laboratories, catalog number: 1422842 )
- Epithelial cell culture medium (see Recipes) (Nowak and Fuchs, 2009)
- Dulbecco’s modified Eagle’s medium/Ham’s F-12 nutrient medium (3:1 Mix) without Calcium (Gibco Invitrogen, special order custom powdered media, catalog number: 90-5010EA)
- Ham’s F-12 nutrient mixture (Thermo Fisher Scientific, GibcoTM, catalog number: 11765054 )
- Penicillin-streptomycin (Thermo Fisher Scientific, GibcoTM, catalog number: 15140148 )
- Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761 )
- L-Glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 21051024 )
- Hydrocortisone (EMD Millipore, catalog number: 3867 )
- Cholera toxin (MP Biomedicals, catalog number: 02150005 )
- Insulin (Sigma-Aldrich, catalog number: I6634 )
- Transferrin (Roche Diagnostics, catalog number: 10652202001 )
- 3,3’,5-Triiodo-L-thyronine (T3) (Sigma-Aldrich, catalog number: T2877 )
- HCl and NaOH for pH adjustment and preparing dilutions of media additives
- Staining buffer (2% FBS) (see Recipes)
Equipment
- Rocking platform (VWR)
- Precision gravity convection incubator set to 37 °C and 5% CO2 (GCA corporation)
- Pipet aid (Drummond) and micro-pipettors (Rainin)
- Refrigerated centrifuge (Eppendorf, model: 5810 R )
- Laminar flow hood (Labgard)
- Autoclave (Steris)
- 4 L beaker (Pyrex)
- 6 L Erlenmeyer flask (Pyrex)
- Glass screw-top bottles, autoclaved (Pyrex)
- Graduated cylinders (Nalgene)
- Stir plate and stir bars (Fisher Scientific)
- pH meter (Fisher Scientific)
- Compressed CO2 source (Praxair)
- Sterile scissors, Iris Ribbon, 4” (Sklar)
- Sterile dissecting forceps, Half-curved 1 mm tip, 4” (George Tiemann & Co)
- FACS Aria II, 4 lasers 405/488/561/633 nm (Becton Dickinson) with 130 μm nozzle (BD, model: FACSAriaTM II )
Procedure
- Dissociation of squamous cell carcinoma into single-cells
- Sacrifice mice according to standard protocol. We use CO2 inhalation followed by cervical dislocation to ensure death has occurred.
- Our tumor-bearing mice contain the eYFP reporter allele (Figure 1A). Because of this, we also sacrifice a healthy, wild-type mouse which does not contain a fluorescent reporter and dissociate keratinocytes from the wild-type anorectal transition zone to obtain cells for unstained and single-stained controls. Dissociate cells from the anorectal transition zone of this healthy, wild-type mouse in parallel to dissociation of tumor cells from the tumor-bearing mouse.
- Carefully dissect the tumor, ensuring that it is separated from skin, fat, muscle and other contaminating tissue (Figure 1A). Remove any necrotic tissue.
- Place the tumor, approximately 0.5 cm2, in 8 ml sterile 1x HBSS in a 10 cm Petri dish and mince using a scalpel until all pieces are uniform and small (Figure 1B).
- Add 100 µl 20% collagenase (see Recipes) to dissociate the minced tumor. Incubate for 45 min while shaking at 37 °C. We place a rocking platform within a dry incubator set at speed 3 rpm.
- After 45 min, the tumor mixture should be homogeneous and may appear viscous (Figure 1C). Add 8.3 µl DNase I (10 mg/ml) to the minced tumor and incubate for an additional 10 min while shaking at 37 °C. After DNAse I treatment, the viscosity of the tumor mixture should be greatly reduced (Figure 1D).

Figure 1. Dissociation of squamous cell carcinoma. A-A’. Keratin14Cre; Tgfbr2 flox/flox mice develop squamous cell carcinoma at the transition between the anal canal and the rectum. We have crossed these mice into mice containing the R26R-eYFPSTOP-flox-STOP allele such that the resulting Keratin14-positive cells, including the anorectal squamous cell carcinoma, express YFP. B. After removing the skin, fat and rectum from the tumor, mince the tumor into small pieces and incubate in HBSS containing 20% collagenase for 45 min (steps A4-A5). C. After 45 min of collagenase treatment, the tumor mixture will appear viscous (step A6). D. After 10 minutes of DNAse I treatment, the viscosity of the tumor mixture will be greatly reduced (step A6). Figures 1B-1D is adapted from McCauley and Guasch, 2013. - Using a 25 ml pipette, add 10 ml cold 1x PBS to the minced tumor and pipette up and down vigorously 8 to 10 times to further dissociate the tumor mixture. Add the minced tumor to a 50 ml conical tube on ice.
- Wash the plate twice with 10-15 ml cold 1x PBS to obtain the maximum number of cells extracted from the tumor and add to the same 50 ml conical tube for each sample dissected.
- Centrifuge for 10 min at 2,000 x g at 4 °C.
- Carefully aspirate the supernatant, as the cell pellet may be very loose at this step. Using a 10 ml pipette, resuspend the cell pellet with 20 ml cold 1x PBS and add 200 µl chelexed FBS.
- Place a 70 µm filter set atop a new 50 ml conical tube. Pre-wet the filter with 1x PBS and filter the tumor cell suspension.
- Place the 70 µm filter in a 60 mm culture dish and add 2 ml of pre-warmed 0.25% trypsin-EDTA to the filter to maximize the number of cells extracted from the minced tumor. Incubate the filter for 10 min at 37 °C. This step is crucial as most of the epithelial CD34+ cells will be obtained at this dissociation step.
- After 5 min, block the activity of the trypsin by adding 5 ml epithelial cell culture media (see Recipes) containing serum to the filter. Mix well the cells with a 5 ml pipette to completely dissociate any clumps. Add the filtered trypsinized cells to the filtered cells in the same 50 ml conical tube from step 11. Wash the 60 mm plate twice with another 5 ml media to maximize the number of cells obtained.
- Place a 40 µm filter set atop a new 50 ml conical tube and pre-wet with epithelial cell culture media. Using a 5 ml pipette, pass the cells through the 40 µm filter.
- Centrifuge for 10 min at 200 x g at 4 °C.
- Aspirate the supernatant and resuspend the cell pellet in 1 ml staining buffer (see Recipes) in PBS to create a single-cell suspension.
- Sacrifice mice according to standard protocol. We use CO2 inhalation followed by cervical dislocation to ensure death has occurred.
- Separation of cancer stem cells from squamous cell carcinoma
- Filter the single-cell suspension through pre-blocked FACS tubes with a cell strainer cap. Pre-wet the cell strainer with PBS before applying the cell suspension to maximize cell number obtained.
Note: To pre-block FACS tubes, we fill each tube that will be used, including those for single-stained and unstained controls, with FBS, allowing the FBS to coat the walls of the tubes. We then decant the FBS and reserve for reuse, and fill each blocked tube with 1x PBS until needed. - Aliquot approximately 100 µl of cell suspension to the appropriate number of pre-blocked FACS tubes for unstained and single-stained controls. For our experiments:
- Wild-type anorectal transition zone cells are split into:
- Unstained control
- 7AAD control (reserve until immediately before sort)
- PE-Cy7 control
- PE control
- Pacific Blue control
- APC control
- YFP+ tumor cells are split into:
- YFP control (100 µl)
- The remainder of the cell suspension (approximately 1 ml) contains all colors and is the sample which will be sorted
- Add the appropriate antibody to each tube.
- Incubate the cells on ice and in the dark for 30 min. Tap the tubes every 5-10 min to disturb the cells from settling at the bottom of the tube.
- Fill each tube with 2% FBS to wash.
- Centrifuge for 5 min at 200 x g.
- Dump the supernatant into a waste container. Approximately 200 µl of buffer, containing the cell pellet, should remain at the bottom of the tube.
- We found that using the biotin-CD34 with the SAV-APC optimized the signal for the rare CD34+ cells within our tumors. For the APC control tube and the sample to be sorted, incubate with secondary antibody SAV-APC for 20 min on ice, protected from light.
Note: Reserve the remainder of the control tubes on ice and protected from light during this time. - Fill these tubes with 2% FBS to wash.
- Centrifuge for 5 min at 200 x g.
- Dump the supernatant into a waste container. Approximately 200 µl of buffer, containing the cell pellet, should remain at the bottom of the tube.
- Resuspend the cell pellet of the tumor sample with 300-800 µl 2% FBS. The volume should be adjusted to account for the size of the cell pellet to optimize flow rate on the cytometer. Keep in mind that a concentrated sample can easily be diluted.
- Bring the 7AAD with the sample to the flow cytometer. We prefer to add the 7AAD to the sample immediately before sorting to minimize cell death due to toxicity. We found 20 µl per million cells to be sufficient to label dead cells.
- Record at least 10,000 events from the unstained and single-color controls to enable compensation.
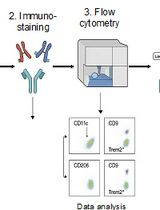
- See Figure 2 for an example of our gating strategy to isolate epithelial CD34+ cancer stem cells. The population hierarchy is:
- Forward scatter (A) versus side scatter (A).
- Side Scatter (H) versus Side Scatter (W) doublet discrimination.
- Forward Scatter (H) versus Forward Scatter (W) doublet discrimination.
- 7AAD negative (live), PE-Cy7 negative (CD11b, CD31, CD45 staining).
- YFP positive.
- PE positive (a6-integrin), Pacific Blue positive (b1-integrin).
- APC negative and positive (CD34).

Figure 2. Gating strategy to isolate epithelial CD34+ cancer stem cells from Tgfbr2-deficient tumors. After dissociation and filtering into a single cell suspension, cells are subjected to fluorescence-activated cell sorting to isolate the rare epithelial CD34+ cancer stem cells. Cells of the appropriate size and granularity are included in P1, and doublets are excluded by the SSC and FSC gates. Live cells are selected by 7AAD exclusion, and CD11b+, CD31+ and CD45+ cells are excluded in the DUMP channel by PE-Cy7 staining. LIVE cells which express YFP under control of the Keratin 14 promoter are then further purified by α6- and β1-integrin expression (PE and Pacific Blue, respectively). Of these YFP+α6+β1+ cells, a pure population of CD34 (APC+) cancer stem cells can then be isolated.
- Forward scatter (A) versus side scatter (A).
- We have successfully isolated live cells for RNA extraction, for returning to culture, and for direct transplantation into recipient mice. Depending on the size of the initial tumor, the efficiency of dissociation, and individual composition of the tumor, we typically obtain between 25,000 and 250,000 live, YFP+, a6-integrin+ b1-integrin+ tumor cells from a tumor approximately 0.5 cm3, and from that tumor population, we typically obtain between 2,000-10,000 CD34+ cancer stem cells.
- For RNA extraction, collect 300 to 10,000 cells directly into 350 µl lysis buffer containing 3.5 µl β-mercaptoethanol in screw-top tubes. Vortex the tubes immediately after sorting and store at -80 °C until RNA extraction. We prefer the QIAGEN RNeasy Micro kit and use the buffer RLT from the kit.
- For return to culture, collect as many cells as possible in 500 µl epithelial cell culture media in a screw-top tube. Centrifuge tubes at 200 x g for 5 min and very carefully aspirate supernatant. Resuspend in epithelial cell culture media and plate. We found that cells did not survive when plating directly on plastic. We prefer to plate cells on irradiated mouse fibroblasts to improve colony formation and survival. We found that plating less than 1,000 cells did not allow for cell survival.
- For direct transplantation, collect as many cells as possible in 500 µl epithelial cell culture media without serum in a screw-top tube. The serum generates bubbles which negatively affect the resuspension in 30% Matrigel. Centrifuge tubes and very carefully aspirate the supernatant. Resuspend in 30% Matrigel, and keep tubes on ice until transplantation (see McCauley and Guasch [2013] for the detailed protocol of the orthotopic transplantation). We have successfully generated tumors by transplanting as few as 100 CD34+ cells. Transplanting larger numbers of cells will accelerate tumor formation.
- For RNA extraction, collect 300 to 10,000 cells directly into 350 µl lysis buffer containing 3.5 µl β-mercaptoethanol in screw-top tubes. Vortex the tubes immediately after sorting and store at -80 °C until RNA extraction. We prefer the QIAGEN RNeasy Micro kit and use the buffer RLT from the kit.
- Filter the single-cell suspension through pre-blocked FACS tubes with a cell strainer cap. Pre-wet the cell strainer with PBS before applying the cell suspension to maximize cell number obtained.
Data analysis
- Because the isolation of rare cells requires inclusion and exclusion of a number of markers, it is imperative to have unstained and single-stained controls for effective compensation of spectral overlap.
- The frequency of epithelial CD34+ cells ranged from 7% to 22%. This is due to the heterogeneity of the squamous cell carcinoma from mouse to mouse. It is therefore recommended to perform at least six biological repeats within each experimental group.
Notes
- All experiments were approved by the Cincinnati Children’s Hospital Research Foundation Institutional Animal Care and Use Committee (protocol number 1D10087) and in agreement with European and national regulation (protocol number 4572), and carried out using standard procedures.
- Trypsinizing the filter is a crucial step that can drastically impact the efficiency of CD34+ cancer stem cells isolated, as the cells may remain in clumps on top of the filter and be unintentionally discarded. Be sure to use pre-warmed 0.25% trypsin-EDTA, to incubate for the full 10:00 at 37 °C, and to vigorously pipet the trypsinized cells to ensure proper dissociation and maximal number of cells obtained.
- It is possible to store the tumor samples in media containing serum overnight at 4 °C and perform the cell isolation the next day. Cell mortality will be increased but the overall percentage of cancer stem cells will not be altered.
- FBS lots need to be tested for optimal growth of the epithelial cells.
- Because epithelial cells are very sensitive to calcium, chelating the FBS is essential.
- While it is possible to collect sorted cells using a 100 μm nozzle, we recommend using a 130 μm nozzle to improve the survival of the large epithelial cells post-sort.
Recipes
- 20% collagenase from Clostridium histolyticum
- Prepare a 20% stock by dissolving 1 g of powdered collagenase in 5 ml 1x sterile phosphate-buffered saline
- Aliquot 250 μl into Eppendorf tubes and store at -20 °C to avoid repeated freezing and thawing that will decrease enzyme activity
- Prepare a 20% stock by dissolving 1 g of powdered collagenase in 5 ml 1x sterile phosphate-buffered saline
- Chelated fetal bovine serum (FBS), prepared as described in (Nowak and Fuchs, 2009)
Day 1- Add 400 g of dry Chelex to a 4 L beaker with a stir bar. Add distilled water to a total volume of 4 L. Cover and stir continuously
- Adjust pH to 7.4 using 10 N HCl. Stir for 20 min, readjust pH with 10 N HCl, and repeat as needed until pH remains stable for more than 20 min
- Place the beaker at 4 °C overnight to allow the Chelex to form a compact pellet
- Carefully decant H2O. Add fresh H2O to 4 L
- Adjust the pH to 7.4 as in Day 1
- Place the beaker at 4 °C for 1 h to allow the Chelex to form a compact pellet
- Carefully decant H2O
- Slowly add two 500 ml bottles of characterized or defined fetal bovine serum to the Chelex
- Stir slowly at 4 °C for 1 h. Try to minimize bubbles
- Place the beaker at 4 °C for 1 h to allow the Chelex to form a compact pellet
- Decant the serum into a 1 L glass bottle and filter the serum through a Nalgene bottle top filter under sterile conditions
- Store unused FBS at 20 °C or use immediately to make E media without calcium
- Add 400 g of dry Chelex to a 4 L beaker with a stir bar. Add distilled water to a total volume of 4 L. Cover and stir continuously
- Epithelial cell culture media, prepared as described in (Nowak and Fuchs, 2009)
- In a 6 L Erlenmeyer flask, combine six packets of Gibco Invitrogen customized DMEM:F12 (3:1) without calcium with distilled water to reach a final volume of 5.5 L
- Add 18.42 g of sodium bicarbonate, 2.85 g of L-glutamine, and 60 ml of 100x penicillin-streptomycin solution
- Adjust pH to 7.2 using 10 N HCl and adjust the volume to 6 L with H2O
- Apply compressed CO2 to the media for 15 min. The media should reach an amber color
- Prepare the following cocktail of media additives:
- 20 ml of 5 mg/ml insulin, dissolved in 0.1 N HCl
- 20 ml of 5 mg/ml transferrin, dissolved in sterile PBS
- 20 ml of 2 x 10-8 M T3, dissolved to 2 x 10-4 in 0.02 N NaOH then further diluted to final concentration in 1x PBS
- 140 ml 1x PBS
- Filter sterilize and store in 37.5 ml aliquots at -20 °C
- Add 75 ml of the above cocktail, 750 ml 10-6 M cholera toxin (dissolved in water), and 750 ml 4 mg/ml hydrocortisone to 1 L of chelated FBS
- Produce final 15% FBS media in 1 L batches by combining 850 ml of the DMEM:F12 media base from step 2 with 150 ml of the supplemented chelated FBS and sterilize using a Nalgene bottle top filter
- Media can be stored in 250 or 500 ml bottles at 20 °C
- In a 6 L Erlenmeyer flask, combine six packets of Gibco Invitrogen customized DMEM:F12 (3:1) without calcium with distilled water to reach a final volume of 5.5 L
- Staining buffer (2% FBS)
1 ml chelexed FBS
49 ml 1x PBS
Note: Make fresh for each use, and keep on ice.
Acknowledgments
The protocol to dissociate the tumor is based on our original protocol published in Methods Mol Biol (McCauley and Guasch, 2013), with minor modifications. The protocol to make chelexed FBS and E media are abbreviated here, but made exactly as described in (Nowak and Fuchs, 2009). An abbreviated description of the flow cytometry protocol appeared in eLife (McCauley et al., 2017). We would like to acknowledge the assistance of the Research Flow Cytometry Core in the Division of Rheumatology at Cincinnati Children’s Hospital Medical Center (supported in part by P30 DK07839) and the Flow Cytometry Core at the CRCM, FRANCE. The protocol related to this work was supported by grants from the V Foundation, the Sidney Kimmel Foundation and in part from the Foundation ARC pour la recherche sur le cancer (GG).
References
- Baumann, M., Krause, M. and Hill, R. (2008). Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer 8(7): 545-554.
- Bonnet, D. and Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3(7): 730-737.
- de Sousa e Melo, F., Kurtova, A. V., Harnoss, J. M., Kljavin, N., Hoeck, J. D., Hung, J., Anderson, J. E., Storm, E. E., Modrusan, Z., Koeppen, H., Dijkgraaf, G. J., Piskol, R. and de Sauvage, F. J. (2017). A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 543(7647): 676-680.
- Guasch, G., Schober, M., Pasolli, H. A., Conn, E. B., Polak, L. and Fuchs, E. (2007). Loss of TGFβ signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell 12(4): 313-327.
- Kreso, A. and Dick, J. E. (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14(3): 275-91.
- Locke, M., Heywood, M., Fawell, S. and Mackenzie, I. C. (2005). Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res 65(19): 8944-8950.
- Malanchi, I., Peinado, H., Kassen, D., Hussenet, T., Metzger, D., Chambon, P., Huber, M., Hohl, D., Cano, A., Birchmeier, W. and Huelsken, J. (2008). Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 452(7187): 650-653.
- McCauley, H. A., Chevrier, V., Birnbaum, D. and Guasch, G. (2017). De-repression of the RAC activator ELMO1 in cancer stem cells drives progression of TGFβ-deficient squamous cell carcinoma from transition zones. Elife 6.
- McCauley, H. A. and Guasch, G. (2013). Serial orthotopic transplantation of epithelial tumors in single-cell suspension. Methods Mol Biol 1035: 231-245.
- Nassar, D. and Blanpain, C. (2016). Cancer stem cells: Basic concepts and therapeutic implications. Annu Rev Pathol 11: 47-76.
- Nowak, J. A. and Fuchs, E. (2009). Isolation and culture of epithelial stem cells. Methods Mol Biol 482: 215-232.
- Prince, M. E., Sivanandan, R., Kaczorowski, A., Wolf, G. T., Kaplan, M. J., Dalerba, P., Weissman, I. L., Clarke, M. F. and Ailles, L. E. (2007). Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A 104(3): 973-978.
- Shimokawa, M., Ohta, Y., Nishikori, S., Matano, M., Takano, A., Fujii, M., Date, S., Sugimoto, S., Kanai, T. and Sato, T. (2017). Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 545(7653): 187-192.
- Wang, J. C. (2010). Good cells gone bad: the cellular origins of cancer. Trends Mol Med 16(3): 145-151.
Article Information
Copyright
![]() McCauley and Guasch. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
McCauley and Guasch. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- McCauley, H. A. and Guasch, G. (2017). Isolation and Separation of Epithelial CD34+ Cancer Stem Cells from Tgfbr2-deficient Squamous Cell Carcinoma. Bio-protocol 7(17): e2524. DOI: 10.21769/BioProtoc.2524.
- McCauley, H. A., Chevrier, V., Birnbaum, D. and Guasch, G. (2017). De-repression of the RAC activator ELMO1 in cancer stem cells drives progression of TGFβ-deficient squamous cell carcinoma from transition zones. Elife 6.
Category
Cancer Biology > Cancer stem cell > Tumor formation > Self-renewal
Cancer Biology > Cancer stem cell > Cell biology assays > Cell isolation and culture
Cell Biology > Cell isolation and culture > Cell isolation > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.