- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Quantitative Heterokaryon Assay to Measure the Nucleocytoplasmic Shuttling of Proteins

Published: Vol 8, Iss 17, Sep 5, 2018 DOI: 10.21769/BioProtoc.2472 Views: 9745
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Many proteins appear exclusively nuclear at steady-state but in fact shuttle continuously back and forth between the nucleus and the cytoplasm. For example, nuclear RNA-binding proteins (RBPs) often accompany mRNAs to the cytoplasm, where they can regulate subcellular localization, translation and/or decay of their cargos before shuttling back to the nucleus. Nucleocytoplasmic shuttling must be tightly regulated, as mislocalization of several RBPs with prion-like domains such as FUS and TDP-43 causes the cytoplasmic accumulation of solid pathological aggregates that have been implicated in neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Traditionally, interspecies heterokaryon assays have been used to determine whether a nuclear protein of interest shuttles; those assays are based on the fusion between donor and recipient cells from two different species (e.g., mouse and human), which can be distinguished based on different chromatin staining patterns, and detecting the appearance of the protein in the recipient nucleus. However, identification of heterokaryons requires experience and is prone to error, which makes it difficult to obtain high-quality data for quantitative studies. Moreover, transient overexpression of fluorescently tagged RBPs in donor cells often leads to their aberrant subcellular localization. Here, we present a quantitative assay where stable donor cell lines expressing near-physiological levels of eGFP-tagged RBPs are fused to recipient cells expressing the membrane marker CAAX-mCherry, allowing to readily identify and image a large number of high-confidence heterokaryons. Our assay can be used to measure the shuttling activity of any nuclear protein of interest in different cell types, under different cellular conditions or between mutant proteins.
Keywords: RNA-binding proteinBackground
To understand the various functions of a protein, it is important to find out where it localizes within cells. Standard microscopic and biochemical methods only reveal the presence of a protein when its steady-state concentration is above the detection threshold. They do not rule out the possibility that it plays additional, important roles where it localizes only transiently (Gama-Carvalho and Carmo-Fonseca, 2001). For example, many RBPs perform functions in different cellular compartments where they accompany their bound mRNAs (often going undetected) and connect multiple steps in eukaryotic gene expression (Müller-McNicoll and Neugebauer, 2013). SR proteins (SRSF1 to SRSF12) are a family of RBPs that regulate transcription, pre-mRNA splicing, 3’end processing and mRNP packaging in the nucleus and appear exclusively nuclear at steady state (Howard and Sanford, 2015; Jeong, 2017). However, most family members shuttle continuously (but to different extents) between the nucleus and the cytoplasm, performing additional functions in mRNA export and translation (Caceres et al., 1998; Sapra et al., 2009; Maslon et al., 2014; Müller-McNicoll et al., 2016; Botti et al., 2017). Changes in RBP shuttling have been described in viral infections, early development, cellular differentiation and neurodegenerative diseases such as ALS and FTD, where pathological accumulation of prion-like RBPs such as FUS and TDP-43 in the cytoplasm forms solid neurotoxic aggregates (Ederle and Dormann, 2017; Liu et al., 2017). Thus, it is very important to know whether an RBP normally shuttles between the nucleus and the cytoplasm and if so, under which circumstances and how it is controlled.
With the tools currently available, it has been difficult to study the cytoplasmic functions of nuclear RBPs and to compare their shuttling abilities. An ingenious method was developed almost thirty years ago–the interspecies heterokaryon assay–in which donor and recipient cells from two different species (e.g., mouse and human) are fused and a protein present only in the donor nuclei gradually appears in the recipient nuclei if it shuttles (Borer et al., 1989). However, this assay provides only qualitative information. The fusion events are identified based on phase-contrast images and donor and recipient nuclei are identified based on distinct chromatin features, which makes the assay laborious, subjective and produces only small numbers of high-confidence heterokaryons. Moreover, fluorescently tagged RBPs are often expressed from transiently transfected plasmids, which results in very different RBP levels in different cells, ranging from barely detectable to non-physiologically high expression that may lead to partially aberrant cytoplasmic or subnuclear localization of RBPs [(Maharana et al., 2018) our unpublished observations]. Altogether, these limitations preclude any comparative analyses.
Here, we present a detailed experimental protocol to perform quantitative shuttling assays in cultured mammalian cells (Figure 1). Our assay is an improvement of the classical heterokaryon assay, with two novelties and a standardized imaging pipeline to perform quantitative measurements. The first novelty is the use of recipient cell lines expressing a fluorescently tagged membrane marker (CAAX-mCherry), which greatly facilitates the identification of heterokaryons containing both a donor and a recipient nucleus and thus allows the rapid and easy identification of a large number of high-confidence heterokaryons. The second novelty is the use of stable clonal donor cell lines, where a fluorescently (eGFP) tagged RBP of interest is expressed from a bacterial artificial chromosome (BAC), which has been integrated into the genome (Botti et al., 2017; Poser et al., 2008). Subsequent clonal selection of cells ensures equal and near-physiological levels of tagged RBPs in every donor cell to facilitate image acquisition, analysis and comparisons.
Our assay has been successfully applied to compare the shuttling activities of different SR proteins in the same cell line, between different cell lines and between differentiation states. Moreover, it allowed us to study the requirements for the shuttling of individual SR proteins using mutated proteins and knockdowns of nuclear export factors (Botti et al., 2017). We have used various cell lines as either donor or recipient cells: these comprise mouse (P19 and NIH3T3) and human (HeLa) cells. Although other cell lines remain to be tested, we are confident that any adherent cell line in which fluorescent RBPs can be expressed at physiological levels is suitable for our assay. This should include primary cells obtained from transgenic animals expressing a fluorescently tagged RBP of interest. We have successfully used P19 cells differentiated into neural cells as donors in our assays (Botti et al., 2017), and it should be possible to study and quantify shuttling of RBPs in other cellular models of differentiation, for example in mouse embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs), or to compare shuttling of RBPs in distinct cellular differentiation fates (Hammarskjold and Rekosh, 2017). Moreover, our assay should allow to quantify changes in shuttling during viral infections and cellular stress, or to assess the impact of disease mutations in RBPs. In principle, our assay could even be adapted to visualize shuttling of long-noncoding RNAs (lncRNAs), for example through the insertion of binding sites for fluorescent MS2 binding protein (MS2-BP) or by inserting an aptamer sequence that binds a fluorescent dye (Ouellet, 2016).
Materials and Reagents
- 10 μl filter tips long (SARSTEDT, catalog number: 70.1116.210 )
- 20 μl filter tips (SARSTEDT, catalog number: 70.760.213 )
- 300 μl filter tips (SARSTEDT, catalog number: 70.765.210 )
- 1,000 μl filter tips (SARSTEDT, catalog number: 70.762.211 )
- 10-cm cell culture dishes (VWR, Thermo Fisher Scientific, catalog number: 734-2043 )
- Cloning discs, size 5 mm (Sigma-Aldrich, SP Scienceware - Bel-Art Products - H-B Instrument, catalog number: Z374458-100EA )
- Disposable Glass Pasteur Pipettes 150 mm (VWR, catalog number: 612-1701 )
- Serological pipettes, 2 ml (VWR, Corning, catalog number: 734-1690 )
- Serological pipettes, 5 ml (VWR, Corning, catalog number: 734-1737 )
- 15-ml Centrifuge tubes (Corning, catalog number: 430791 )
- 2-ml microcentrifuge tubes (SARSTEDT, catalog number: 72.691 )
- 1,000 µl pipette tips ART® 1000E Barrier Tips (Thermo Fisher Scientific, catalog number: 2079E )
- 12-well plates for cell culture (VWR, Thermo Fisher Scientific, catalog number: 734-2156 )
- Precision coverslips, 18 mm, borosilicate glass 0.17 ± 0.005 mm (Carl Roth, catalog number: LH23.1 )
- Microscope slides (VWR, catalog number: 631-1550 )
- Bacterial artificial chromosome (BAC) containing the gene encoding an eGFP-tagged RBP of interest (Botti et al., 2017 and Poser et al., 2008; see Notes 1-3)
- Plasmid for expression of fluorescent plasma membrane marker of a different color (e.g., CAAX-mCherry, plasmid TH0477, Stewart et al., 2011; Botti et al., 2017; see Note 4)
- DMEM, high glucose, GlutaMAXTM Supplement, pyruvate (Thermo Fisher Scientific, catalog number: 31966047 )
- Fetal Bovine Serum (Thermo Fisher Scientific, catalog number: 10270106 )
- Penicillin-Streptomycin (10,000 U/ml) (Thermo Fisher Scientific, catalog number: 15140122 )
- Puromycin 10 mg/ml (Thermo Fisher Scientific, GibcoTM, catalog number: A1113803 )
- Geneticin® Selective Antibiotic (G418 Sulfate) (50 mg/ml) (Thermo Fisher Scientific, catalog number: 10131035 )
- Trypsin 0.05% EDTA (Thermo Fisher Scientific, catalog number: 25300054 )
- Dulbecco's Phosphate Buffered Saline (Sigma-Aldrich, catalog number: D8537 )
- Gelatin solution bioreagent 2% in H2O (Sigma-Aldrich, catalog number: G1393 )
- Cycloheximide solution 100 mg/ml in DMSO (Sigma-Aldrich, catalog number: C4859 )
- Polyethylene Glycol (PEG) 1500 in 75 mM HEPES (Roche Diagnostics, catalog number: 10783641001 )
- Phosphate buffered saline 10x (Sigma-Aldrich, catalog number: P5493 )
- Pierce 16% Formaldehyde (w/v), Methanol-free (Thermo Fisher Scientific, catalog number: 28908 )
- 30.Water, Molecular Biology Reagent (Sigma-Aldrich, catalog number: W4502 )
- ProLong® Diamond Antifade Mountant (Thermo Fisher Scientific, InvitrogenTM, catalog number: P36970 )
- TWEEN® 20 (Sigma-Aldrich, catalog number: P7949 )
- Tris (Carl Roth, catalog number: 4855.2 )
- Hoechst 34580 (Sigma-Aldrich, catalog number: 63493 )
- Trizma® base (Sigma-Aldrich, catalog number: T1503 )
- Sodium Chloride (NaCl), BioXtra (Sigma-Aldrich, catalog number: S7653 )
- Hydrochloric acid 36.5-38.0% (HCl), for molecular biology (Sigma-Aldrich, catalog number: H1758 )
- DMEM containing 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin (see Recipes)
- PBS containing 0.1% gelatin (see Recipes)
- 10x TBS (see Recipes)
- TBST (see Recipes)
- 4% Formaldehyde in 1x PBS (see Recipes)
- Hoechst 34580 stock solution (1 mg/ml) (see Recipes)
- TBST containing 0.25 μg/ml Hoechst 34580 (see Recipes)
Equipment
- Hemocytometer
- Timer
- Fume hood
- Cell culture hood (Thermo Fisher Scientific, model: HerasafeTM KSP , type KSP 12)
- Fine tip curved tweezers
- P10 pipettor (VWR, catalog number: 613-5259 )
- P20 pipettor (VWR, catalog number: 613-5260 )
- P200 pipettor (VWR, catalog number: 613-5263 )
- P1000 pipettor (VWR, catalog number: 613-5265 )
- Mini vacuum pump, KNF (A. Hartenstein, catalog number: AP86 )
- Fluid aspiration system (VACCUBRAND, model: VHCpro )
- CO2 incubator (Thermo Fisher Scientific, model: HeracellTM 150i )
- Vortex mixer (VWR, catalog number: 444-1372 )
- Pipette controller (accu-jet® pro, Brand, catalog number: 26300 )
- -20 °C freezer
- Refrigerator
- Inverted microscope (Motic, model: AE31 )
- Confocal laser-scanning microscope (ZEISS, model: LSM 780 )
Software
- Fiji (ImageJ Version 2.0.0-rc-43/1.51h or more recent)
- Microsoft Excel (version 14.6.7 or more recent)
Procedure
See Figure 1 for an overview of the quantitative heterokaryon shuttling assay.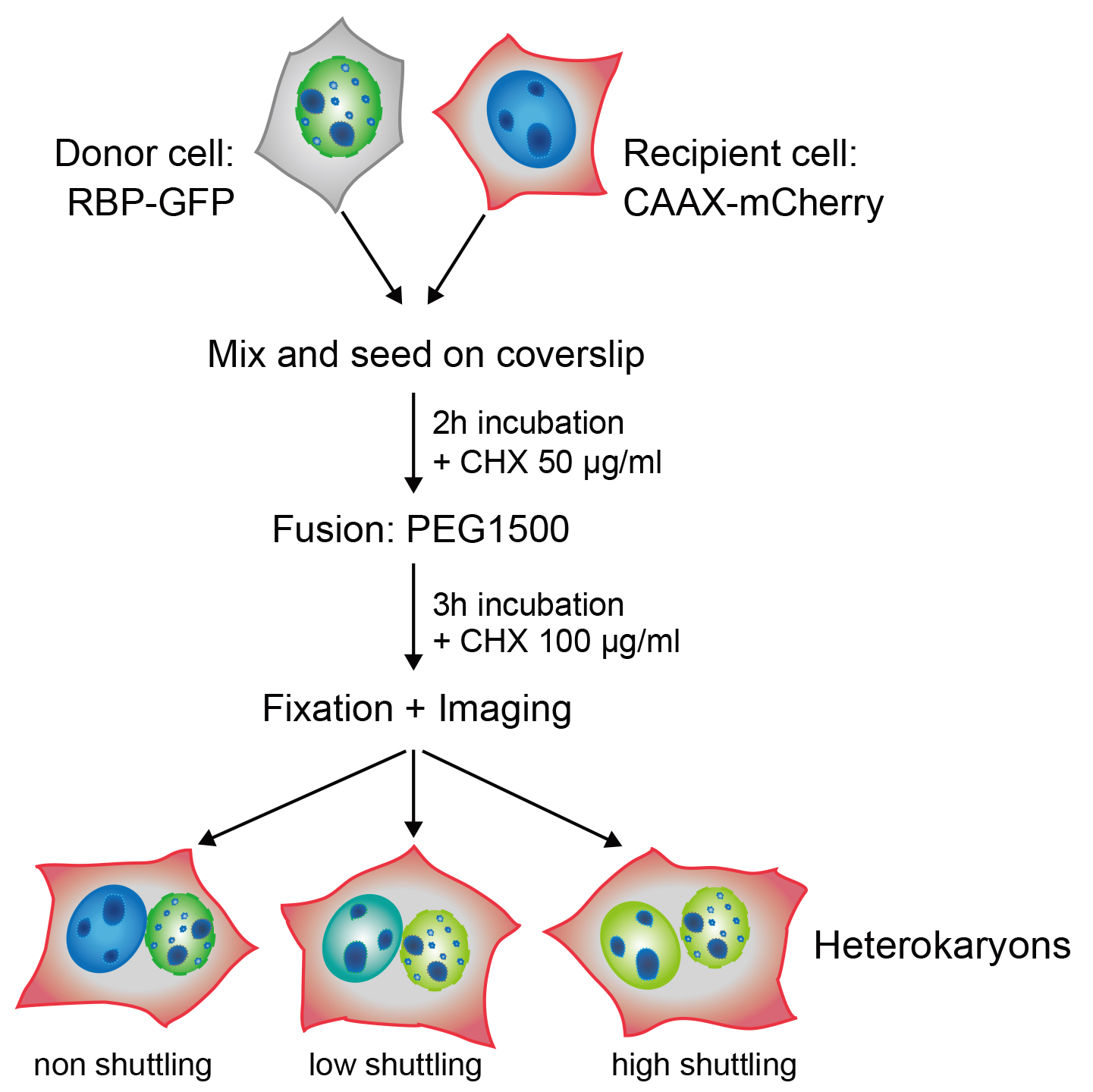
Figure 1. Overview of the quantitative heterokaryon shuttling assay. Donor cells expressing a GFP-tagged nuclear RBP are seeded together with recipient cells expressing CAAX-mCherry tethered to their cell membranes. Cells are fused with PEG1500 in the presence of cycloheximide to block new protein synthesis, i.e., to avoid appearance of the RBP in the common cytoplasm not due to shuttling. After 3 h, cells are fixed and imaged by confocal microscopy. Heterokaryons suitable for analysis can be readily identified as they contain only two nuclei, at least one of which is green, and both are surrounded by a red membrane. Nuclei are imaged by confocal microscopy and GFP fluorescence is quantified in both the donor and the recipient nucleus. Given that shuttling is allowed to occur in the presence of a protein synthesis inhibitor (cycloheximide), any fluorescence in the recipient nucleus is due to shuttling of the RBP.
- Generate clonal transgenic donor and recipient cell lines
- Generate a donor cell line expressing an eGFP-tagged RBP of interest from a BAC randomly integrated into the genome as described in Botti et al. (2017) and Poser et al. (2008) (see Notes 1-3).
- Generate a recipient cell line expressing a plasma membrane-localized marker of a different color (e.g., CAAX-mCherry) from a plasmid randomly integrated into the genome as described in Botti et al. (2017) (see Note 4).
- Using fluorescence activated cell sorting (FACS) or cloning discs, isolate clones with different fluorescence levels for the donor cell line and at least one clone with high fluorescence for the recipient cell line (see Note 5).
- If a specific antibody against the RBP of interest is available, estimate by Western blotting the expression levels of the eGFP-tagged RBP in different clones and select a clone with near-endogenous levels.
- Prepare individual cultures of donor and recipient cells
Passage donor and recipient cells onto separate 10-cm culture dishes to obtain nearly confluent cultures 1-3 days later. - Mix and seed donor and recipient cells (Timing: 30 min) (see Note 6)
- For each combination of donor and recipient cells, place 4 sterile 18-mm coverslips into different wells of a 12-well plate. If using cell lines that normally require a pre-coated surface to increase their adherence, pre-coat the coverslips accordingly. For example, P19 cells require coating with PBS containing 0.1% gelatin, and we normally pre-coat the coverslips for 24 h at 37 °C when using this cell line.
- Estimate the confluence (e.g., 50%, 75%, 100%) of both donor and recipient cells.
- Aspirate medium and gently wash with 5 ml of pre-warmed sterile PBS. Aspirate the PBS thoroughly and add 1.5 ml of 0.05% Trypsin. Incubate for 3-5 min at RT or 37 °C (optimal conditions must be determined for each cell line).
- Help the cells detach from the dishes by tilting the plate in every direction.
- Add 3 ml of appropriate medium (e.g., DMEM) containing 10% FCS to inhibit trypsin activity.
Critical Step: Disrupt the cell clumps by pipetting up and down 20 times in the cell culture dish using the fastest speed setting on your pipet controller. Make sure you obtain single cell suspensions of both donor and recipient cells by visual inspection under the microscope. - Transfer each single cell suspension to a 15-ml tube.
- Calculate the concentration of both donor and recipient cell suspensions using a standard method (e.g., using a hemocytometer). Alternatively, use approximate values according to your cell line, culture dish (i.e., surface area) and confluence. For example, a 10-cm dish with HeLa cells grown at confluence contains approximately 8.8 x 106 cells, and following Steps C3 to C6, the corresponding cell suspension (in a volume of 4.5 ml) would contain approximately 2.0 x 106 cells/ml.
- In a 2-ml tube, prepare 2-ml of a 1:1 donor:recipient mixed cell suspension containing approximately 1 x 106 to 2 x 106 cells from each cell suspension. For example, if the donor cell culture was approximately 50% confluent (~4.4 x 106 cells per dish) and the recipient cell culture was approximately 100% confluent (~8.8 x 106 cells per dish), mix 1.333 ml of donor cell suspension (~9.8 x 105 cells/ml) with 0.667 ml of recipient cell suspension (~2.0 x 106 cells/ml) for a total of approximately 2.6 x 106 cells.
- Vortex the mixed cell suspension for 10 sec at medium speed.
- Transfer 1 ml of the mixed cell suspension to a 2-ml tube containing 1 ml of medium (2-fold dilution). Vortex for 10 sec at medium speed. This and the subsequent dilutions should help to obtain the optimal density for cell fusion and imaging (see Procedure I).
- Similarly, perform two more 2-fold serial dilutions (corresponding to 4-fold and 8-fold dilutions). The 8-fold dilution should contain approximately 2.5 x 105 to 5.0 x 105 cells.
- Transfer 1 ml of each mixed cell suspension (undiluted, 2-fold, 4-fold and 8-fold dilutions) onto a coverslip in a 12-well plate prepared at Step C1. Make sure the coverslips are not floating at the surface of the medium. If some of them do, help them sink at the bottom using a sterile tip. If the coverslips were pre-coated (e.g., with PBS containing 0.1% gelatin), aspirate the coating solution from each well just before adding the mixed cell suspension.
- Attachment of cells to coverslips (Timing: overnight [8-16 h])
Incubate at 37 °C, 5% CO2 for 8-16 h.
Critical Step: Incubate long enough to allow the cells to attach to the coverslips but briefly enough to avoid cell division. Recently divided cells readily fuse together, often resulting in heterokaryons containing more than two nuclei, which should not be used for shuttling quantification (see Procedure I and J). For example, mouse P19 cells divide every 16 h in the exponential growth phase, and we incubate for a maximum of 8 h at this step when using this cell line (see Note 6). - Inhibition of protein synthesis (Timing: 2.5 h) (see Note 7)
Critical Step: Some cell lines might not be tightly attached to the coverslips at this stage. To prevent the loss of cells, be very careful when performing each medium change or wash. This advice should be followed until the cells are fixed (Step H3). Always add medium or buffer on the sidewall of the well, not directly onto the coverslip.- Aspirate medium and gently add 1 ml of medium containing 50 μg/ml cycloheximide (CHX). Incubate for 2 h at 37 °C with 5% CO2.
- Aspirate medium and gently add 1 ml of medium containing 100 μg/ml CHX. Incubate for 30 min at 37 °C with 5% CO2.
- Cell fusion (Timing: 30 min) (see Note 8)
- Aspirate medium and gently add 1 ml of pre-warmed PBS containing 100 μg/ml CHX.
- Start the timer for 2 min and 10 sec. Then, one coverslip at a time, aspirate PBS/CHX and gently add 0.25 ml of pre-warmed PEG 1500.
Critical Step: Proceed swiftly (approx. 10 sec) from coverslip to coverslip. Each coverslip should be incubated for 2 min (± a few seconds). Hold the fluid aspiration device with one hand and the pipette controller with the other. For aspiration, use the same glass Pasteur pipette for all samples. To distribute PEG 1500, use the same 2-ml pipette for all samples. We do not recommend processing more than 12 coverslips within one batch. - Without removing the PEG 1500, gently add 3 ml of PBS to each coverslip. Maintain a pace of about 10 sec per coverslip.
- Aspirate PBS and perform two more washes with 3 ml of PBS each.
- Aspirate PBS and gently add 1 ml of medium containing 100 μg/ml CHX.
- Optional: Briefly visually inspect the overall fusion efficiency under the microscope. Many cells containing several nuclei can be observed at this stage.
- Shuttling (Timing: 3 h)
Incubate for 3 h at 37 °C with 5% CO2 (see Note 9). - Cell fixation and preparation of microscope slides (Timing: 1.5 h, overnight)
- Aspirate medium containing 100 μg/ml CHX and add 1 ml of ice-cold PBS containing 100 μg/ml CHX.
- Perform a second wash with 1 ml of ice-cold PBS containing 100 μg/ml CHX.
- One coverslip at a time, aspirate PBS containing 100 μg/ml CHX and gently add 0.5 ml of ice-cold PBS containing 4% formaldehyde. Protect from light and incubate at RT for 20 min.
Caution: Formaldehyde is toxic and should be handled in a fume hood. - Wash twice each with 1 ml of PBS.
- Remove PBS and add 0.5 ml of TBST containing 0.25 μg/ml Hoechst 34580. Protect from light and incubate at RT for 30 min.
Caution: Hoechst 34580 is potentially carcinogenic, so precautions should be taken for handling and disposal. - Remove TBST containing 0.25 μg/ml Hoechst 34580 and wash twice each with 1 ml of PBS. Leave the coverslips in the PBS from the second wash.
- Using fine curved tweezers, retrieve each coverslip, remove as much PBS as possible by touching a napkin with the edge of the coverslip, and transfer (facing up) onto a sheet of paper. Label each coverslip by writing on the sheet of paper next to it.
- Protect from light and allow to dry completely (at least 15 min).
- One coverslip at a time, add 13.5 μl of ProLong® Diamond Antifade mounting reagent to a microscope slide, pick up the coverslip using tweezers and invert it onto the drop of mounting reagent.
Critical: Avoid pipetting air bubbles onto the slide. - Protect from light and incubate at RT for at least 16 h on a level surface.
Pause Point: Slides may be stored up to 24 h at RT and then for several weeks at 4 °C until imaging.
- Imaging (Timing: 4-6 h per sample for 15 heterokaryons)
Note: We recommend that the imaging settings (excitation/emission wavelengths, laser power, gain, offset, etc.) be optimized for each donor cell line (e.g., expressing an eGFP-tagged protein of interest) and recipient cell line (e.g., expressing CAAX-mCherry) grown separately on coverslips and processed in parallel with the samples. Make sure that the combination of fluorophores and settings do not generate any cross-talk.- For each sample (i.e., for each pair of donor-recipient cell lines), choose the coverslip containing the optimal cell density by visual inspection of Hoechst fluorescence. Cell density should be high enough so that pairs of nuclei often appear in close proximity to each other but clearly separated from surrounding nuclei (see Notes 10-12).
- Move the objective toward the periphery of the coverslip until cell density is reduced and/or cells appear damaged, then move the objective back into the region suitable for analysis (see Note 13).
- Perform a systematic search for the desired cell fusion (donor-recipient) events using Hoechst fluorescence. Nuclei in fused cells are very close to each other and often display complementary shapes, although it is not always the case (see examples in Figures 2 and 3). When using cells from two different species (e.g., mouse and human), finding heterokaryons can sometimes be facilitated by the presence of distinct heterochromatin patterns specific to each species (Borer et al., 1989). Note that heterokaryons suitable for analysis (i.e., clearly containing only one donor and one recipient nuclei) constitute rare events (< 0.01% of the cells on the examined coverslip).
- Before imaging, confirm that both nuclei are surrounded by a common, red membrane in the mCherry channel (see Note 14).
- Using the confocal microscope acquisition settings, find the optimal focus (middle of the cells on the Z-axis), define the area to be imaged and take a picture in all three channels (mCherry, eGFP and Hoechst, see Figures 2 and 3). Use the same frame size and resolution for all pictures in a given analysis; according to our experience, imaging an area of 67.4 x 67.4 μm with a resolution of 1,024 x 1,024 pixels generates high-quality images with a reasonable scanning time and almost always allows us to include at least one nucleus from an unfused recipient cell (or at least part of it) for background subtraction.
- Acquire z-stacks of eGFP and Hoechst fluorescence with the same number of slices for all pictures in a given analysis. In our experience, 10 to 12 slices with an interval of 0.31 μm allow covering almost the entire nucleus in the Z-axis. To minimize scanning time and bleaching of eGFP fluorescence, we recommend imaging first all slices in the eGFP channel and then all slices in the Hoechst channel. We do not deem it necessary to acquire z-stacks of CAAX-mCherry as it significantly increases scanning time; however, make sure to keep the picture generated in Step I5) as evidence that the two nuclei originate from a fused donor-recipient cell.
- Resume the systematic search for desired donor-recipient cell fusion events and repeat Steps I4 to I6. We recommend imaging a minimum of 15 pairs of nuclei per sample for proper statistical analysis.
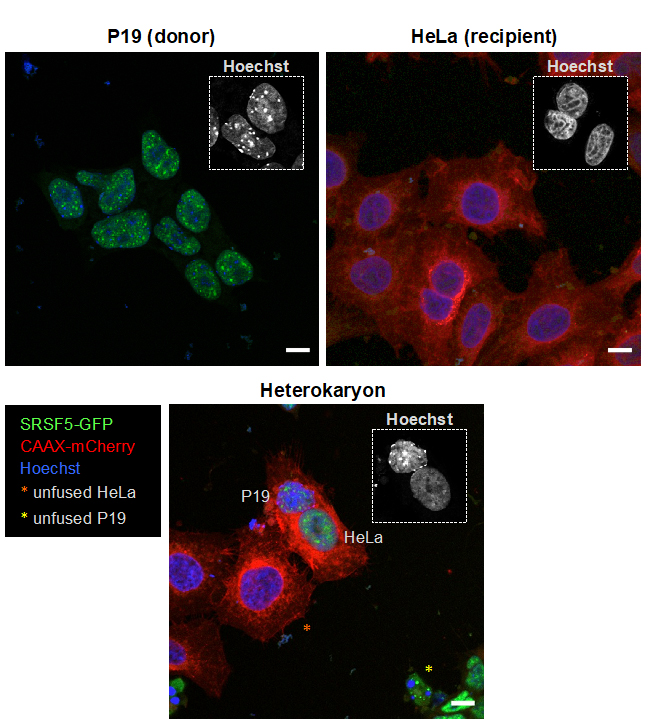
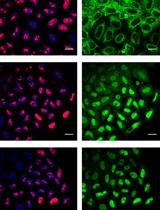
Figure 2. Examples of clonal donor and recipient cell lines that can be used in quantitative heterokaryon assays. Upper left panel, mouse P19 cells expressing GFP-tagged SRSF5, an RBP with nucleocytoplasmic shuttling activity (Botti et al., 2017), are used as donor cells. SRSF5-GFP is expressed at near endogenous levels from a BAC randomly integrated into the genome, resulting in physiological localization of the protein throughout the nucleoplasm with enrichment in nuclear speckles. Note the highly similar GFP fluorescence levels in all cells due to the use of a clonal cell line. An inset shows the characteristic Hoechst staining pattern for mouse P19 cells. Upper right panel, human HeLa cells expressing CAAX-mCherry in their cell membranes are used as recipient cells. CAAX-mCherry is expressed at uniform levels in all recipient cells due to the selection of a high-expressing clone following random integration of plasmid TH0477 into the genome. An inset shows the characteristic Hoechst staining pattern for HeLa cells. Lower panel, donor and recipient cell lines shown in upper panels were used in the quantitative heterokaryon assay. The image shows a heterokaryon, i.e., a cell containing one nucleus from a donor P19 cell and one nucleus from a recipient HeLa cell, both surrounded by a common membrane marked with CAAX-mCherry. An inset shows the Hoechst staining patterns of both nuclei of the heterokaryon. The orange asterisk indicates an unfused HeLa cell and the yellow asterisk, an unfused P19 cell. Note the conspicuous presence of GFP signal in the recipient HeLa nucleus and its absence from the unfused HeLa nucleus (see also Figure 3). Also note the absence of CAAX-mCherry signal in membranes surrounding unfused P19 nuclei. Scale bars = 10 μm.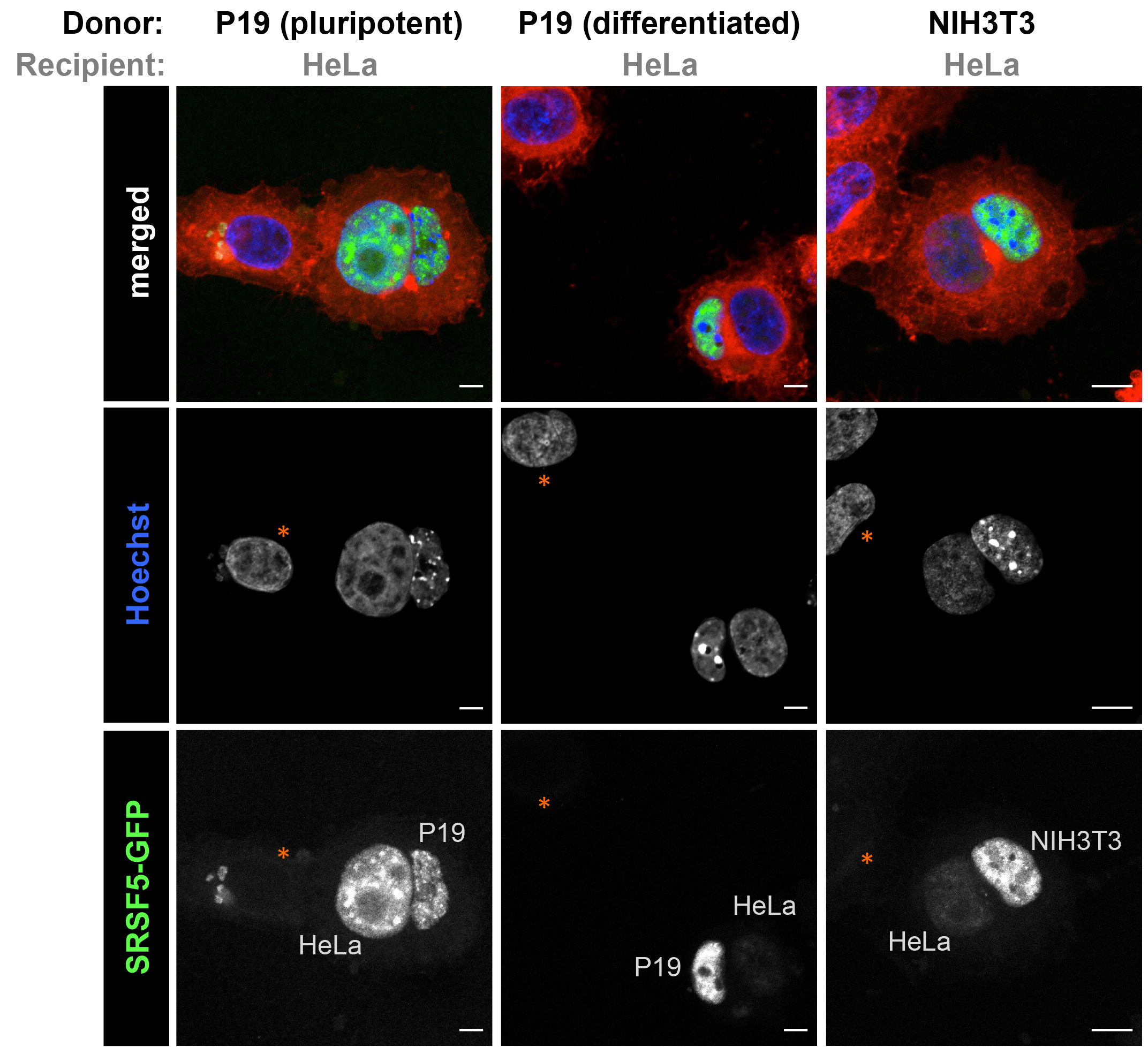
Figure 3. Cell type-specific shuttling activity can be studied using quantitative heterokaryon assays. Left and middle panels, mouse P19 cells expressing SRSF5-GFP were used as donor cells before (left) and after (middle) 9 days of differentiation into neural cells. Right panel, a mouse NIH3T3 clonal cell line expressing SRSF5-GFP was used as donor cell line. SRSF5-GFP shuttles efficiently in pluripotent (P19) cells but poorly in differentiated (P19 and NIH3T3) cells. See Botti et al. (2017) for quantification and comparison of the shuttling activity of SRSF5 in these different cell types. Note the complete absence of GFP signal from unfused HeLa nuclei (indicated by orange asterisks). Scale bars: left panel = 5 µm; middle panel = 5 µm; right panel = 10 μm.
- Image analysis and quantification of fluorescence (Timing: 2-3 h for 15 images)
- Open Fiji (ImageJ Version 2.0.0-rc-43/1.51h or more recent).
- Open the Region Of Interest (ROI) Manager by selecting “Analyze → Tools → ROI Manager…”.
- Open image (select “View stack with: Hyperstack”, “Color mode: Composite” and “Autoscale”.
- Select “Image → Stacks → Z Project…”, “Start slice: 1”, “Stop slice: 10” (or 12, include all slices), “Projection type: Max Intensity”.
- Select “Image → Color → Split Channels”.
- Click on the picture generated in the Hoechst channel and select “Image → Lookup Tables → Grays”.
- Select “Image → Adjust → Brightness/Contrast” and make both the donor and recipient nuclei as visible as possible while still being able to distinguish them from each other.
- If the donor and recipient nuclei are in very close proximity to each other, draw a line between them using the Pencil Tool.
- Adjust the brightness and contrast to see the entire donor and recipient nuclei as clearly as possible.
- Zoom on the donor/recipient nuclei as much as possible so that they are entirely visible in the window. Using Freehand selections, draw the border of the donor cell.
- In the ROI Manager window, click “Add [t]”. This will generate an ROI with a specific number.
- In the list, click on the number corresponding to the ROI and select “Rename…” and then “Rename As: Image 1 donor”.
- Draw the border of the recipient nucleus, add to the ROI Manager and rename as “Image1 recipient”.
- Zoom out as necessary and zoom in on the nucleus of an unfused recipient cell.
- Draw the border of the nucleus from the unfused recipient cell, add to the ROI Manager and rename as “Image 1 background”.
- Click on the original (.czi) picture and select “Image → Stacks → Z Project…”, “Start slice: 1”, “Stop slice: 10” (or 12, include all slices), and “Projection type: Sum Slices” (see Note 15).
- Select “Image → Color → Split Channels.
- Click on the image of the eGFP fluorescence and select “Image → Type → 16-bit”.
- In the ROI Manager window, select the ROI corresponding to the donor nucleus and click on “Measure”. A Results window will open. Make sure the following data have been generated: Area, Mean, Min, Max and Slice.
- In the ROI Manager window, select the ROI corresponding to the recipient nucleus and click on “Measure”.
- Repeat for the background nucleus.
- Close all images and repeat Steps J3 to J21 with image 2.
- To save the ROIs for documentation, select in the ROI Manager “More >>… Save”.
- Save the results in a Microsoft Excel file by clicking on the Results window and selecting “File → Save As…”.
- Open the Excel File. For each image, subtract the mean pixel intensity (Mean) of the unfused recipient nucleus from that of both the donor and recipient nuclei. This yields the background subtracted mean pixel intensity for each nucleus.
- For both the donor and recipient nuclei, multiply the background subtracted mean pixel intensity by the area to obtain the total pixel intensity.
- Divide the total pixel intensity of the recipient nucleus by that of the donor nucleus and multiply by 100 to obtain the value of the Shuttling capacity (%).
Data analysis
- To compare the shuttling of a protein in two different conditions or the shuttling of two different proteins, we recommend imaging a minimum of 15 heterokaryons per condition or protein from at least two independent experiments (e.g., 7-8 heterokaryons per coverslip from two independent experiments).
- The mean shuttling activity of a protein in a given condition should be calculated from at least 15 heterokaryon images.
- We apply Student's t-tests and paired Wilcoxon rank-sum tests to test whether the shuttling activity is significantly different between the different conditions or proteins. For this, all quantified and background subtracted shuttling capacity values are assembled into columns with the protein name as header into a .txt file. Plots and tests are performed in R.
Example:
read.table("/path/Quantification_HKA.txt", head=TRUE, sep="\t") -> experiment_HKA
summary(experiment_HKA)
boxplot(experiment_HKA, las = 2, ylab = "Shuttling capacity HKA)", boxfill=c("lightgrey"))
wilcox.test(experiment_HKA $Control, experiment_HKA $Mutant, paired=TRUE)
Notes
- Protein tagging in bacterial artificial chromosomes (BACs) and subsequent generation of transgenic cell lines has been described in detail in (Poser et al., 2008).
- The readout of shuttling assays is the re-distribution of a protein from a donor to a recipient nucleus. In the original assay (Borer et al., 1989), the emergence of proteins of interest in recipient nuclei (from a different species) was qualitatively assessed by indirect immunofluorescence using species-specific monoclonal antibodies; however, for some proteins, specific antibodies are difficult–if not impossible–to generate. To circumvent this, cells have been transiently transfected with epitope-tagged cDNAs encoding proteins of interest prior to fusion with recipient cells (Pinol-Roma and Dreyfuss, 1991; Caceres et al., 1998; Lin et al., 2005). However, transient expression from plasmids often leads to overexpression, which in turn can affect the functions and subcellular distribution of RBPs (Maharana et al., 2018; our unpublished observations). Moreover, RBP levels will vary substantially from cell to cell, precluding the identification of comparable heterokaryons. To overcome these limitations, we strongly recommend using clonal cell lines stably expressing fluorescently tagged proteins from bacterial artificial chromosomes (BACs) that have been integrated into the genome in single copy (Poser et al., 2008; Botti et al., 2017). The large size of BACs ensures the presence of all gene regulatory elements (e.g., promoters, enhancers, intronic sequences and untranslated regions), which preserves endogenous ratios of splice isoforms and results in expression levels similar to those of the endogenous gene (Figure 2, Änkö et al., 2012; Müller-McNicoll et al., 2016; Botti et al., 2017). Alternatively, fluorescent tags can be introduced into RBPs expressed from their endogenous loci using genome editing by CRISPR/Cas9 (Van Nostrand et al., 2017).
- We also recommend using control cell lines in each experiment. We have used cells expressing eGFP-tagged PRPF8 (a spliceosomal component) and the nuclear export factor (NXF1) as negative and positive controls for shuttling, respectively (Botti et al., 2017). Other RBPs for which shuttling has been confirmed or ruled out can be used. However, one should bear in mind that shuttling of a given RBP might vary depending on the cell type and/or differentiation state (Figure 3, Botti et al., 2017; Hammarskjold et al., 2017).
- To readily identify true heterokaryons, our assay requires the use of a membrane marker that is expressed only in recipient cells. We have successfully used clonal recipient cell lines expressing CAAX-mCherry from a plasmid randomly integrated into their genomes (TH0477, Stewart et al., 2011; Botti et al., 2017). CAAX is an isoprenylated peptide which is tethered to cell membranes (Wright and Philips, 2006). However, other membrane markers might be equally suitable. Stable transgenic cell lines using plasmids can be generated in essentially the same way as when using BACs. See Note 1.
- Our assay is based on quantitative cell imaging, which requires a standardized imaging protocol with identical settings (laser power, gain, digital offset, etc.) for acquisition of all images. To achieve this, we strongly recommend the use of clonal lines for both donor and recipient cell lines, preferentially selected for endogenous expression level of the studied RBP. Clonal cell lines can be obtained either by FACS sorting, which requires access to a specialized core facility or the help of a qualified technician, or without any specialized equipment or expertise using cloning discs.
- The most critical part of the protocol: To produce reliable results, only heterokaryons containing one donor and one recipient nucleus should be quantified. To increase the frequency of examinable heterokaryons–and thereby reduce microscopy time/increase statistical power–experimental conditions should be optimized for different combinations of donor-recipient cell lines in the following three ways.
First, use an appropriate ratio of donor to recipient cells. In principle, a 1:1 ratio should be used (and is a good starting point) to increase the probability that heterokaryons contain one donor and one recipient nucleus. However, different cell types adhere more or less tightly to glass coverslips, and given that several medium changes/washes must be carried out prior to cell fusion, this ratio might have to be optimized according to the combination of cells used. In addition, some cell lines have a faster growth rate than others, and a compromise should be made between strong attachment and low number of cell division, as recently divided cells tend to fuse together and reduce the frequency of examinable heterokaryons. For example, when we use rapidly dividing mouse P19 cells as donors and HeLa cells as recipients, we allow a shorter (6 to 8 h) time for adhesion than when using mouse NIH3T3 cells with the same recipients (12-16 h) to avoid P19 cell division, but we use a P19:HeLa cell ratio of 2:1 or 3:1 as P19 cells adhere less quickly and strongly than HeLa and NIH3T3 cells to the coverslips and many P19 cells are lost during the washes. When using cell types that require coating (e.g., with gelatin, laminin, etc.), we recommend pre-incubating the coverslips for a sufficient time, preferably several hours at 37 °C, before seeding the cells.
Second, it is crucial to obtain a single-cell suspension of both donor and recipient cells prior to mixing and seeding. This might require pipetting up and down harshly to break the cell clumps. Do not hesitate to sacrifice viability of some cells to obtain true single-cell suspensions.
Third, use an optimal cell density. To obtain heterokaryons, cells should adhere to the coverslip close enough to each other so that their membranes can fuse upon chemical treatment (see below); however, cell suspensions should be diluted enough to avoid multiple fusion events. To find the best compromise between single and multiple fusions, we recommend performing serial 2-fold dilutions of a mixed (donor-recipient) cell suspension prior to seeding and determining by microscopy the optimal dilution to search for suitable heterokaryons. Although the use of recipient cell lines expressing a fluorescent membrane marker greatly facilitates the identification of heterokaryons appropriate for analysis, finding clear fusion events between a single donor and a single recipient cell can remain challenging, and it is worth investing some time to determine the optimal dilutions with which to proceed to the search. - Nuclear proteins are synthesized in the cytoplasm. To determine whether a nuclear protein shuttles to the cytoplasm and back to the nucleus, it is therefore essential to block protein synthesis before and during the shuttling assay. In this way, any newly emerging fluorescent protein in a recipient nucleus should originate from the donor nucleus, not from the pool of newly synthesized proteins in their common cytoplasm. A confirmed non-shuttling nuclear RBP (e.g., splicing factor PRPF8; Sapra et al., 2009) should always be used as a negative control; the absence of shuttling confirms efficient protein synthesis inhibition. We have used two different inhibitors of protein synthesis (cycloheximide and puromycin) yielding essentially similar results (Botti et al., 2017).
- Polyethylene glycol (PEG) is water soluble, so it can be easily removed by washing; however, a prolonged incubation can be toxic for most cell lines (our observations). It is important to use the same incubation time for all samples in a comparative shuttling analysis.
- Following cell fusion, sufficient time should be allowed in order to detect and measure the shuttling of an RBP of interest. Several factors can affect the shuttling efficiency of a given protein. We have shown that different members of the SR protein family shuttle at different rates, which correlated with the length of their phosphorylatable RS domain (Botti et al., 2017). Shuttling rates can differ greatly depending on the biological functions of the RBP. For example, whereas some nuclear proteins such as nucleolin appear to slowly “leak” from to the cytoplasm, requiring about 24 h to be detected in a recipient nucleus, shuttling of nucleocytoplasmic transport factors can be detected within minutes (Gama-Carvalho et al., 2001) and reach equilibrium (i.e., 50% in both donor and recipient nuclei) within 3 h (Caceres et al., 1998; Sapra et al., 2009; Botti et al., 2017). When comparing the shuttling efficiencies of different RBPs, it is crucial to fix the cells before they reach equilibrium. Moreover, in order to minimize variation, the exact same incubation time should be allowed for shuttling in each replicate of the analysis.
Another factor that can affect shuttling efficiency is the combination of donor and recipient nuclei. For example, if a given protein is exported from the donor nucleus but not from the recipient nucleus, the protein might get trapped and accumulate in the recipient nucleus, and the shuttling capacity of the protein might appear greater than it is in reality, sometimes reaching over 50% (unpublished observation). In such a case, it might be necessary to reduce the shuttling time to be able to quantify differences (e.g., between mutants and WT proteins).
Finally, the cell cycle stage of donor and recipient nuclei might, in some cases, affect shuttling capacity of a protein under study. The nuclear volume and the number of NPCs almost double during interphase in dividing cells (Maeshima et al., 2011). A larger nucleus should, in theory, allow a faster import and a slower export of shuttling proteins due to its larger surface area being in contact with the cytoplasm and its smaller surface area being in contact with the nucleoplasm. We have observed that larger recipient nuclei tend to harbor higher total fluorescence levels (unpublished observation); however, such an effect does not significantly affect the results when a sufficient number of heterokaryons are imaged. Nevertheless, the shuttling of some proteins might vary throughout the cell cycle, and in some cases it might be useful to synchronize the cells (e.g., in G1) before fusion. - If none of the coverslips shows an optimal density, adjust dilutions in subsequent experiments.
- Some cell fusion events might seem to happen only between cells of the same cell line (donor or recipient cell line). One possible reason is the faster growth rate of either donor or recipient cells. If one cell line grows too quickly, try to reduce the attachment time at Step D1.
- In some cases, although a donor:recipient cell ratio of 1:1 has been used at Step C8, one cell type (either donor or recipient) appears to predominate on the slides. One possible reason is that different cell types attach more or less firmly to coverslips, and either donor or recipient cells might be lost at a higher rate during medium changes and washes.
- Cells at the periphery of the coverslip are sometimes not suitable for imaging/analysis: according to the Hoechst fluorescence, their nuclei might appear smaller, brighter and irregularly shaped, and their specific chromatin features might be less apparent. The fluorescence levels of such cells tend to be lower in both the eGFP and mCherry channels. These are regions containing presumably damaged cells.
- Acquiring Z-stacks, which are necessary for proper quantification, is time consuming. The use of recipient cells with a fluorescent marker in their membranes (e.g., CAAX-mCherry) allows to readily distinguish true heterokaryons from donor/recipient cells that are merely in close proximity but not fused. If a quick look under the microscope suggests that it is a true heterokaryon, proceed to imaging. Otherwise, resume the search for suitable heterokaryons.
- Even if care has been taken to avoid pixel saturation during imaging, the sum of slices will make some of the pixels appear saturated. This should affect only a minority of the pixels in the region of interest, and in our experience it doesn’t significantly impact the results. However, if one suspects that too many pixels appear saturated in the sum of slices and that this might affect the analysis, fluorescence should be quantified in separate slices and then summed.
Recipes
- DMEM containing 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin
- Add 50 ml of heat-inactivated FBS to 500 ml of DMEM
- Add 5 ml of Penicillin-Streptomycin (10,000 U/ml)
- Mix well and store at 4 °C (keep sterile)
- PBS containing 0.1% gelatin
- Add 25 ml of 2% gelatin to 500 ml of sterile 1x PBS
- Mix well and store at 4 °C (keep sterile)
- 10x TBS (1 L)
- Dissolve 24 g of Trizma® base and 88 g of NaCl in 900 ml of dH2O
- Adjust pH to 7.6 with HCl
- Adjust volume to 1 L and verify pH
- Store at RT
- TBST (1 L)
- Add 100 ml of 10x TBS to 900 ml of dH2O
- Add 0.5 ml of Tween® 20
- Mix well and store at RT
- 4% Formaldehyde in 1x PBS (40 ml, for 80 coverslips)
- In a 50-ml tube, add 4 ml of 10x PBS to 26 ml of H2O (Sigma)
- In a fume hood, add 10 ml of 16% formaldehyde
- Mix by vortexing for 10-15 sec
- Store at -20 °C in 4-ml aliquots (each enough for 8 coverslips)
- Hoechst 34580 stock solution, 1 mg/ml (5 ml)
- Dissolve the entire contents of the vial (5 mg) in 5 ml of H2O (Sigma)
- Store in aliquots at -20 °C and protect from light
- Hoechst 34580, 0.25 μg/ml in TBST (4 ml, for 8 cover slips)
- In a 15-ml tube, add 1 μl of 1 mg/ml Hoechst 34580 to 4 ml of H2O (Sigma)
- Mix by vortexing for 10-15 sec
- Prepare fresh, keep on ice and protect from light
Acknowledgments
This work was supported by funding from the German Research Foundation (DFG) to MMM (CEF-MC and SFB902). We are grateful to M. C. Steiner for developing the first version of the assay and K. M. Neugebauer for inspiration and guidance. We thank H. Schewe (FCAM) for support on confocal microscopes. This protocol was adapted from procedures published in Botti et al. (2017).
Competing interests
The authors declare no competing financial interests.
References
- Änkö, M. L., Muller-McNicoll, M., Brandl, H., Curk, T., Gorup, C., Henry, I., Ule, J. and Neugebauer, K. M. (2012). The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol 13(3): R17.
- Borer, R. A., Lehner, C. F., Eppenberger, H. M. and Nigg, E. A. (1989). Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56(3): 379-390.
- Botti, V., McNicoll, F., Steiner, M. C., Richter, F. M., Solovyeva, A., Wegener, M., Schwich, O. D., Poser, I., Zarnack, K., Wittig, I., Neugebauer, K. M. and Müller-McNicoll, M. (2017). Cellular differentiation state modulates the mRNA export activity of SR proteins. J Cell Biol 216(7): 1993-2009.
- Caceres, J. F., Screaton, G. R. and Krainer, A. R. (1998). A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev 12(1): 55-66.
- Ederle, H. and Dormann, D. (2017). TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett 591(11): 1489-1507.
- Gama-Carvalho, M. and Carmo-Fonseca, M. (2001). The rules and roles of nucleocytoplasmic shuttling proteins. FEBS Lett 498(2-3): 157-163.
- Gama-Carvalho, M., Carvalho, M. P., Kehlenbach, A., Valcarcel, J. and Carmo-Fonseca, M. (2001). Nucleocytoplasmic shuttling of heterodimeric splicing factor U2AF. J Biol Chem 276(16): 13104-13112.
- Hammarskjold, M. L. and Rekosh, D. (2017). SR proteins: To shuttle or not to shuttle, that is the question. J Cell Biol 216(7): 1875-1877.
- Howard, J. M. and Sanford, J. R. (2015). The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip Rev RNA 6(1): 93-110.
- Jeong, S. (2017). SR Proteins: Binders, regulators, and connectors of RNA. Mol Cells 40(1): 1-9.
- Lin, S., Xiao, R., Sun, P., Xu, X. and Fu, X. D. (2005). Dephosphorylation-dependent sorting of SR splicing factors during mRNP maturation. Mol Cell 20(3): 413-425.
- Liu, E. Y., Cali, C. P. and Lee, E. B. (2017). RNA metabolism in neurodegenerative disease. Dis Model Mech 10(5): 509-518.
- Maeshima, K., Iino, H., Hihara, S. and Imamoto, N. (2011). Nuclear size, nuclear pore number and cell cycle. Nucleus 2(2): 113-118.
- Maharana, S., Wang, J., Papadopoulos, D. K., Richter, D., Pozniakovsky, A., Poser, I., Bickle, M., Rizk, S., Guillen-Boixet, J., Franzmann, T., Jahnel, M., Marrone, L., Chang, Y. T., Sterneckert, J., Tomancak, P., Hyman, A. A. and Alberti, S. (2018). RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360(6391): 918-921.
- Maslon, M. M., Heras, S. R., Bellora, N., Eyras, E. and Caceres, J. F. (2014). The translational landscape of the splicing factor SRSF1 and its role in mitosis. Elife: e02028.
- Müller-McNicoll, M. and Neugebauer, K. M. (2013). How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat Rev Genet 14(4): 275-287.
- Müller-McNicoll, M., Botti, V., de Jesus Domingues, A. M., Brandl, H., Schwich, O. D., Steiner, M. C., Curk, T., Poser, I., Zarnack, K., and Neugebauer, K. M. (2016). SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev 30: 553-566.
- Ouellet, J. (2016). RNA Fluorescence with light-up aptamers. Front Chem 4: 29.
- Pinol-Roma, S. and Dreyfuss, G. (1991). Transcription-dependent and transcription-independent nuclear transport of hnRNP proteins. Science 253(5017): 312-314.
- Poser, I., Sarov, M., Hutchins, J. R., Heriche, J. K., Toyoda, Y., Pozniakovsky, A., Weigl, D., Nitzsche, A., Hegemann, B., Bird, A. W., Pelletier, L., Kittler, R., Hua, S., Naumann, R., Augsburg, M., Sykora, M. M., Hofemeister, H., Zhang, Y., Nasmyth, K., White, K. P., Dietzel, S., Mechtler, K., Durbin, R., Stewart, A. F., Peters, J. M., Buchholz, F. and Hyman, A. A. (2008). BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods 5(5): 409-415.
- Sapra, A. K., Änkö, M. L., Grishina, I., Lorenz, M., Pabis, M., Poser, I., Rollins, J., Weiland, E. M. and Neugebauer, K. M. (2009). SR protein family members display diverse activities in the formation of nascent and mature mRNPs in vivo. Mol Cell 34(2): 179-190.
- Stewart, M. P., Helenius, J., Toyoda, Y., Ramanathan, S. P., Muller, D. J. and Hyman, A. A. (2011). Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 469(7329): 226-230.
- Van Nostrand, E. L., Gelboin-Burkhart, C., Wang, R., Pratt, G. A., Blue, S. M. and Yeo, G. W. (2017). CRISPR/Cas9-mediated integration enables TAG-eCLIP of endogenously tagged RNA binding proteins. Methods 118-119: 50-59.
- Wright, L. P. and Philips, M. R. (2006). Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res 47(5): 883-891.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- McNicoll, F. and Müller-McNicoll, M. (2018). A Quantitative Heterokaryon Assay to Measure the Nucleocytoplasmic Shuttling of Proteins. Bio-protocol 8(17): e2472. DOI: 10.21769/BioProtoc.2472.
- Botti, V., McNicoll, F., Steiner, M. C., Richter, F. M., Solovyeva, A., Wegener, M., Schwich, O. D., Poser, I., Zarnack, K., Wittig, I., Neugebauer, K. M. and Müller-McNicoll, M. (2017). Cellular differentiation state modulates the mRNA export activity of SR proteins. J Cell Biol 216(7): 1993-2009.
Category
Molecular Biology > Protein > Protein shuttling
Cell Biology > Cell-based analysis > Nucleocytoplasmic shuttling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.