- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Ex vivo Whole-cell Recordings in Adult Drosophila Brain



Published: Vol 8, Iss 14, Jul 20, 2018 DOI: 10.21769/BioProtoc.2467 Views: 8640
Reviewed by: Steven BoeynaemsAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Jan 2006
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Cost-effective and efficient, the fruit fly (Drosophila melanogaster) has been used to make many key discoveries in the field of neuroscience and to model a number of neurological disorders. Great strides in understanding have been made using sophisticated molecular genetic tools and behavioral assays. Functional analysis of neural activity was initially limited to the neuromuscular junction (NMJ) and in the central nervous system (CNS) of embryos and larvae. Elucidating the cellular mechanisms underlying neurological processes and disorders in the mature nervous system have been more challenging due to difficulty in recording from neurons in adult brains. To this aim we developed an ex vivo preparation in which a whole brain is isolated from the head capsule of an adult fly and placed in a recording chamber. With this preparation, whole cell recording of identified neurons in the adult brain can be combined with genetic, pharmacological and environmental manipulations to explore cellular mechanisms of neuronal function and dysfunction. It also serves as an important platform for evaluating the mechanism of action of new therapies identified through behavioral assays for treating neurological diseases. Here we present our protocol for ex vivo preparations and whole-cell recordings in the adult Drosophila brain.
Keywords: Adult brain dissectionBackground
The fruit fly (Drosophila melanogaster) has been used to make key discoveries in a variety of fundamental areas in neuroscience including learning and memory (Bolduc et al., 2008; Cervantes-Sandoval et al., 2016), synapse formation and regulation (Genç et al., 2017), and circadian rhythms (Allada et al., 1998; Guo et al., 2016). Mutants identified through both forward and reverse genetic screens have also provided useful models of human neurological disorders including Fragile X syndrome, Parkinson’s Disease, Huntington Disease and epilepsy disorders (Pallos et al., 2008; Parker et al., 2011; Liu et al., 2012; Sears and Broadie, 2017). Much of what we have learned in this research comes from electrophysiological recordings and calcium imaging at the neuromuscular junction (NMJ), from neurons in dissociated primary culture, or from the central nervous system of embryos and larvae. Though these methods have been instrumental in our understanding, to elucidate the underlying cellular mechanisms of neurological processes in adult animals, it is important to have electrophysiological access to individual neurons of the adult brain.
Recordings from neurons in the adult CNS were made possible by development of two complementary systems in the mid-2000s. One involves exposing and desheathing a small area of brain making it possible to obtain intracellular recordings from neurons in a live, behaving adult fly (Wilson et al., 2004; Hige et al., 2015; Nagel and Wilson, 2016). This preparation is best suited to recording from populations of neurons on the dorsal surface of the brain. The second preparation involves removing the whole brain from the adult head capsule and placing it in a recording chamber (Gu and O’Dowd, 2006 and 2007). This provides access to neurons in the entire brain and allows for easy environmental manipulations. Although the process is invasive and may cause damage to the brain, intact neurons and functional circuits can be persevered and maintained for up to one hour after skillful dissection. Labs have used whole brain dissection and whole-cell recordings to characterize the electrical properties of circadian neurons (Sheeba et al., 2008b), uncover the electrical cellular mechanisms responsible for sleep and arousal (Sheeba et al., 2008a), discover a new light-sensing pathway in the brain (Ni et al., 2017), determine the mechanism of action for a common pesticide (Qiao et al., 2014), find a memory suppressor miRNA that regulates an autism susceptibility gene (Guven-Ozkan et al., 2016), and describe synaptic dysfunction in a model of Parkinson’s Disease (Sun et al., 2016).
Our lab uses this protocol extensively to study the cellular mechanisms of genetic epilepsy associated with mutations in SCN1A, a gene that encodes NaV1.1 sodium channels that are highly expressed in inhibitory, GABAergic neurons in the human brain. Using homologous recombination, and more recently CRISPR/Cas9 mediated gene editing, we have introduced specific SCN1A missense mutations into the same location in the Drosophila sodium channel gene, para. We have shown that all of the mutations causing febrile seizure phenotypes in humans that we have examined (K1270T, S1231R, R1648H/C), also result in heat-induced seizure phenotypes in the adult fly (Sun et al., 2012; Schutte et al., 2014 and 2016). To evaluate how specific mutations alter sodium currents and neuronal activity, we perform electrophysiological analyses of sodium currents in knock-in flies carrying SCN1A mutations, focused primarily on GABAergic, local neurons (LNs) in the antennal lobe. Whole-cell recordings from the cell bodies of LNs can be used to evaluate sodium currents and firing properties in mutant compared to wild-type neurons. The ability to rapidly exchange extracellular recording solutions in the ex vivo preparation allows fast and reversible elevation of the temperature to assess constitutive and temperature-dependent changes in sodium currents and firing properties in knock-in mutant compared to wild-type neurons. Fast perfusion also facilitates evaluation of the acute effects of potential anti-convulsant drugs on sodium currents and firing properties. Here we present our updated protocol for ex vivo whole-cell recordings in adult Drosophila brains, including fly dissection and preparation, data acquisition, and analysis.
Materials and Reagents
Materials
- Ex vivo preparation
- 35 mm Petri dish
- 1cc Plastic syringes
- 27 G ½ needles
- 35 mm Petri dish
- Electrophysiology
Reagents
- Ex vivo preparation
- Sodium chloride (NaCl) (1 M) (Sigma-Aldrich, catalog number: S5886 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P4504 )
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: C4901 )
- Magnesium chloride solution (MgCl2) (1 M) (Sigma-Aldrich, catalog number: M1028 )
- Glucose (Sigma-Aldrich, catalog number: G8270 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- L-cysteine (Sigma-Aldrich, catalog number: C7352 )
- Papain suspension (Worthington Biochemical, catalog number: LS003126 )
- Sodium hydroxide (NaOH) (10 N) (Fisher Scientific, catalog number: S25550 )
- Sodium chloride (NaCl) (1 M) (Sigma-Aldrich, catalog number: S5886 )
- Electrophysiology
- Colbalt (II) chloride (CoCl2) (Sigma-Aldrich, catalog number: 60818 )
- Tetraethylammonium chloride (TEA) (Sigma-Aldrich, catalog number: T2265 )
- 4-aminopyridine (4-AP) (Sigma-Aldrich, catalog number: 275875 )
- (+)-Tubocurarine chloride (curarine) (Tocris Bioscience, catalog number: 2820 )
- Picrotoxin (Sigma-Aldrich, catalog number: P1675 )
- Potassium gluconate (Kgluconate) (Sigma-Aldrich, catalog number: P1847 )
- EGTA (Sigma-Aldrich, catalog number: E4378 )
- Adenosine 5’-triphosphate disodium salt hydrate (Na2ATP) (Sigma-Aldrich, catalog number: A2383 )
- Potassium hydroxide (KOH) (8 N) (Sigma-Aldrich, catalog number: P4494 )
- Cesium hydroxide (CsOH) (50 wt. % in H2O) (Sigma-Aldrich, catalog number: 232068 )
- D-gluconic acid solution (49-53 wt. % in H2O) (Sigma-Aldrich, catalog number: G1951 )
- Colbalt (II) chloride (CoCl2) (Sigma-Aldrich, catalog number: 60818 )
Equipment
- Ex vivo preparation
- AA Forceps
- Fine-tip tweezers
- Osmometer (such as Wescor, model: 5600 )
- Dissecting stereomicroscope (such as Nikon Instruments, model: SMZ800N )
- Gooseneck light source (such as Edmund Optics, model: Fiber-Lite® Illuminator System, catalog number: 35-277 )
- AA Forceps
- Electrophysiology
- Pipette puller (such as NARISHIGE, model: PC-100 )
- Upright microscope (such as OLYMPUS, model: BX51WI )
- Amplifier (such as Molecular Devices, model: Axopatch 200B )
- Digitizer (such as Molecular Devices, model: Digidata 1500B )
- Micromanipulator (such as Sutter Instrument, model: MP-225 )
- Air table and Faraday cage (such as Sutter Instrument, model: AT-3036 )
- Peristaltic pump system (such as Cole-Parmer, catalog number: EW-77910-20 )
- (Additional) In-Line Solution Heater (Harvard Apparatus, model: SHM-828 )
- (Additional) Temperature Controller (Harvard Apparatus, model: CL-100 )
- Pipette puller (such as NARISHIGE, model: PC-100 )
Software
- pClamp software suite (minimum version 9.0)
Procedure
- Ex vivo preparation
Note: Whole-brain dissection from adult fly Video of this section is available on Jove (Gu and O’Dowd, 2007).- Prepare dissecting solution (see Recipe 1). The dissecting solution is best used within 4-5 h after preparation.
- Put fly vial on ice for ~1 min to anesthetize flies (watch carefully–it will only take a short time to slow the flies down, and too long on ice will kill them).
- Place a small drop (~60 μl) of dissecting solution in the lid of 35 mm Petri dish.
- Use a pair of fine-tip tweezers and the dominant hand to pick up an anesthetized fly by the wings or legs.
- With the non-dominant hand, use syringe needle to pin down thorax onto the Petri dish (NOT into the dissecting solution).
- Take a second syringe needle with the dominant hand and decapitate fly.
- Stabilize hands by resting them on microscope stage, and keep fingers close to the needle tips. With head facing up, pin down the fly’s left eye with the needle in the non-dominant hand (Needle 1). With needle in the dominant hand (Needle 2), make a vertical cut just medial to Needle 1 that extends from the rostral to caudal surface (Figure 1).
- Turn brain 180 degrees. Pin down the fly’s right eye with Needle 1, and again with Needle 2 make a vertical cut just medial to Needle 1 that extends from the rostral to caudal surface (Figure 1).

Figure 1. Cutting the cuticle. Pin down each eye and cut off the outer portion. Then pin and cut away the mouthparts. The cuticle will now be open on three sides to dissecting solution. - Pin down mouthparts with Needle 1 and with Needle 2, make a horizontal cut that extends the entire width of the fly head and bisects the cut edges of the eyes (Figure 1).
- Carefully push the cut head into drop of dissecting solution and set a timer for 5 min.
- Turn over the fly head so that the rostral side is on the dish. Pin down the bottom edge of the rostral cuticle, beneath the brain, with Needle 1, and use Needle 2 to open “the flap of” the caudal cuticle like a book (Figure 2A).
- The intact fly brain should now be exposed. Still pinning down the rostral cuticle with Needle 1, use the back side of Needle 2 to gently push the brain out of the head capsule (Figure 2B).
- Clean the brain by pinning down connective tissue with Needle 1 and gently pushing the brain away with Needle 2. As much connective tissue should be removed as possible (Figure 2C).
- Entire dissection process should take less than 10 min.
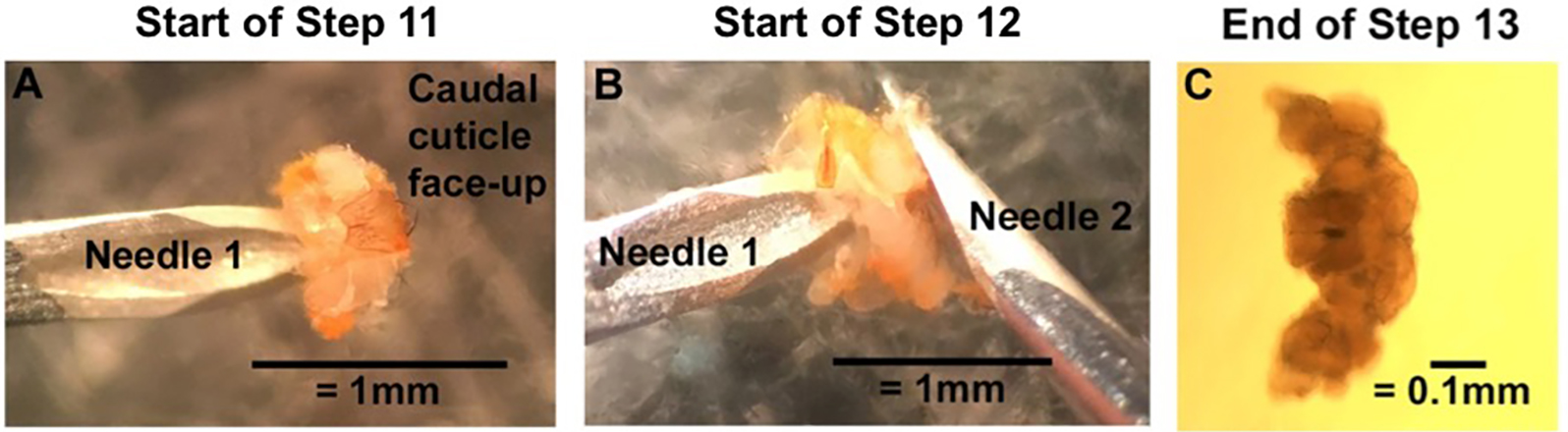
Figure 2. Removing the brain from the cuticle. A. With the caudal cuticle face-up, use Needle 1 to pin down the underhanging tab of the rostral cuticle. Use the backside of Needle 2 to open the caudal cuticle like a book. B. Still pinning the inner surface of the rostral cuticle with Needle 1, use the backside of Needle 2 to push the brain out of the head cuticle. C. Clear off connective tissue to end with a clean and intact Drosophila brain. Clean brain imaged with a compound microscope with the rostral side up. Under a dissecting microscope, the brain will look milky gray with bright white connective tissue.
- Prepare dissecting solution (see Recipe 1). The dissecting solution is best used within 4-5 h after preparation.
- Mounting fly brain for electrophysiology
- Fill recording chamber with external solution. We use a chamber and platform set that allows for solution perfusion as well as heating of the chamber (Figure 3).

Figure 3. Recording chamber and holder. The chamber is connected to a perfusion system on either side to keep solution flowing. The chamber also contains connections to a temperature controller that allows us to heat the chamber directly to warm solution surrounding the brain. A brain holder (described in Step B4) secures the brain for recording. - Transfer the brain to recording chamber using a 200 μl pipet. Note that the fly brain may get stuck on the inside of the pipet tip especially if the connective tissue was not fully cleaned off the brain.
- Use a needle to gently push the brain to the desired position. For most of our experiments, we position the caudal part of the brain on the chamber surface with the antennal lobes facing up. The position may be changed based on the specific need of different experiments.
- Stabilize the brain using a brain holder as shown in Figure 4. The brain holder is made in our lab. The frame is made with a platinum wire with dimensions of 6 mm x 6 mm. The nylon fibers are glued on to the frame with super glue and the distance between each fiber is about 0.5-1 mm to accommodate the variation in brain size (but most intervals between fibers are closer to 0.5 mm). To best stabilize the brain, the fibers should cross the brain at the junction between the optic lobes and central brain region (Figure 4).

Figure 4. Mounting the brain. A. The brain holder is made of a bent platinum wire crossed by horizontal fibers a little less than 0.5 mm apart. B. Orient the dissected brain as shown and hold in place with platinum holder.
- Fill recording chamber with external solution. We use a chamber and platform set that allows for solution perfusion as well as heating of the chamber (Figure 3).
- Whole-cell Recording
Our headstage is on the right of our rig setup and we use borosilicate glass micropipettes (100 μl) that are pulled on a micropipette puller in two stages: final tip resistance of 9-10 Ω.- Circulate external solution with a perfusion (peristaltic pump) system. Oxygenation of the external solution is not necessary when it is constantly perfused at low speed. No minimal speed has been established, but we generally keep the perfusion at 0.8 ml/min for our recording chamber which holds 1-1.5 ml external solution. The circulating speed may vary depending on experimental needs, for example, fast drug administration or temperature change may require more rapid perfusion. However, perfusion that is too rapid may generate electrical noise or flush the brain out of place.
- Fill a pipette with internal solution and load onto headstage. Apply positive pressure to keep the pipette tip free of debris.
- Use manipulator to bring pipette above brain and slightly to the right.
- Local neurons are suspended above the neuropil area of the antennal lobes. As pipette approaches LNs, cells should move away slightly from the pipette (Figure 5). For those who wish to investigate excitatory neurons, cholinergic projection neurons (PNs) are located medially on the antennal lobe. The spikelet and current amplitudes of this population can be quite small. On the dorsal lateral side of the brain, large and small ventral lateral neurons implicated in circadian rhythms and sleep have also been studied using this protocol.
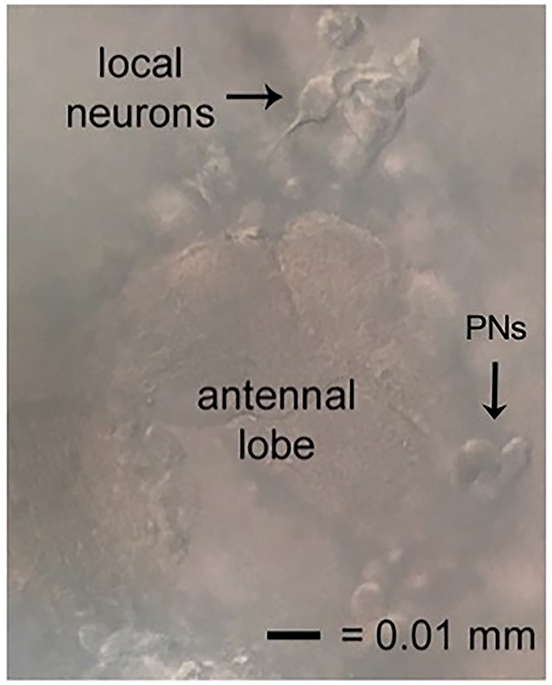
Figure 5. Locating local neurons. Local neurons good for recording will be suspended above, but tethered to, the neuropil of the antennal lobe, have smooth cell membranes, and move away slightly when approached. The excitatory neurons of this circuit, the projection neurons (PNs), also shown. - Release pressure on cell moving away to form a giga-ohm seal. Then break into the cell to record.
Note: Osmolarity is crucial to the success of patches and recordings. If cells instantly break in without forming a giga-ohm seal, the osmolarity difference is too large. If cells don’t move away from the pipette or can’t be broken into, the osmolarity difference is too small. The external solution should be 15-20 mOsm L-1 greater than the internal solution. We use a vapor pressure osmometer to approximate, but adjustments are common. A 3% dilution decreases osmolarity by about 10 mOsm L-1. We recommend keeping the internal solution in a small sealed container, such as 1.5 ml centrifuge tubes. If the internal solution in an open container, the osmolality can change drastically (20-30 mOsm L-1) in a few hours due to water evaporation.
- Circulate external solution with a perfusion (peristaltic pump) system. Oxygenation of the external solution is not necessary when it is constantly perfused at low speed. No minimal speed has been established, but we generally keep the perfusion at 0.8 ml/min for our recording chamber which holds 1-1.5 ml external solution. The circulating speed may vary depending on experimental needs, for example, fast drug administration or temperature change may require more rapid perfusion. However, perfusion that is too rapid may generate electrical noise or flush the brain out of place.
Data analysis
Record and analyze traces using the pClamp suite. Sample traces for our protocols are shown below (Figures 6-8). More details regarding data processing and analysis can be found in Sun et al., 2012 and Schutte et al., 2014.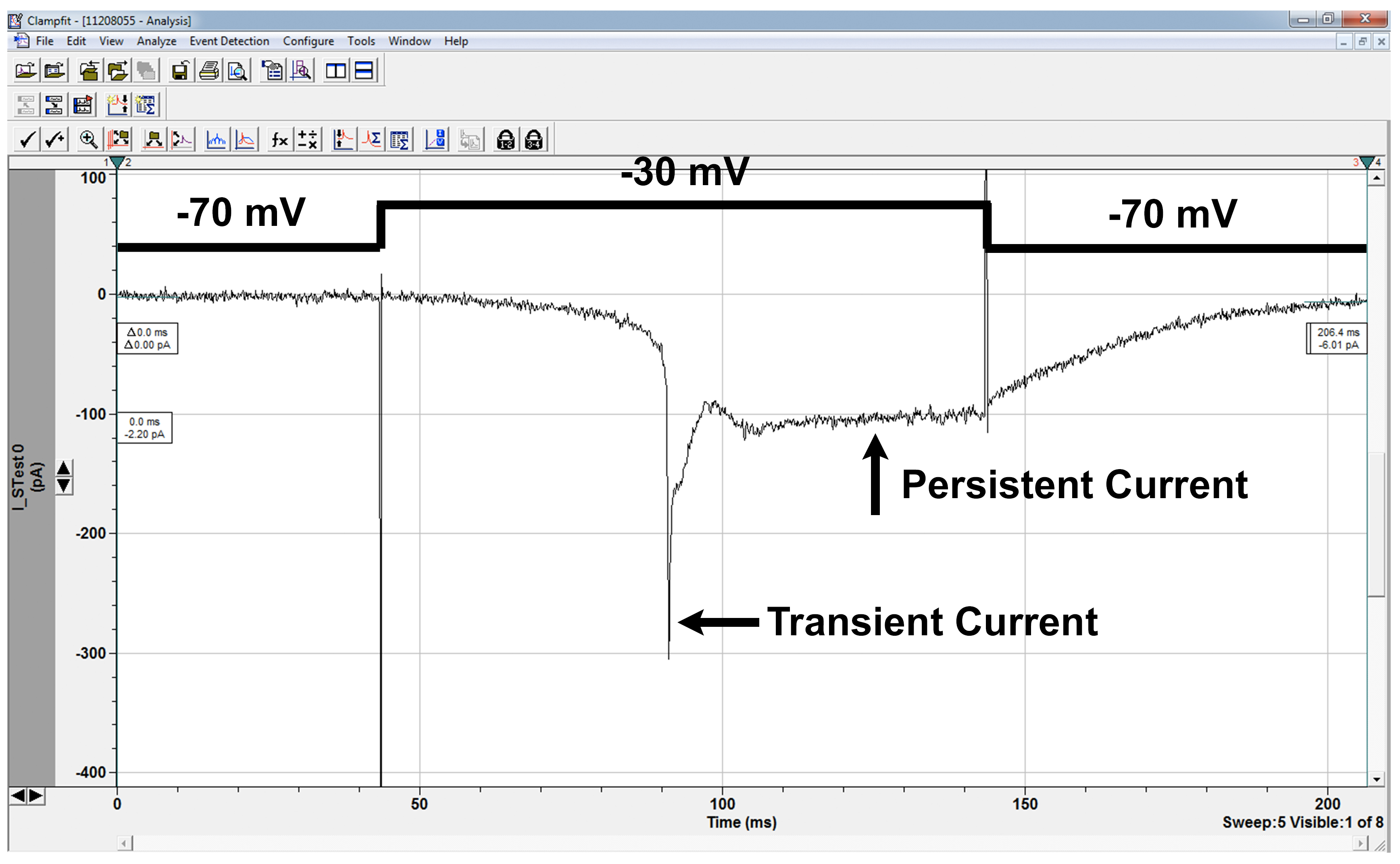
Figure 6. Sodium currents. Sodium current in LN in response to depolarizing voltage-step as shown. Sodium currents are not well space-clamped as the sodium channels are located on the processes, not the cell body we record from, and this is also responsible for the delay between the depolarizing step and the onset of channel opening. In this trace, the cell was held at -70 mV and a series of depolarizing traces applied to activate the channel. -30 mV was the first sweep to activate the currents, and is characterized as the activation threshold. The transient current amplitude is about 300 pA, and the persistent current amplitude is about 100 pA in this case. Mutant cells may have activation thresholds and/or current amplitudes greater or lower than wildtype cells. The channel deactivates after the stimulus step has ended, a phenotype that is sometimes impaired in mutant cells. Voltage sweeps can be applied after channel activation to investigate channel deactivation threshold as well.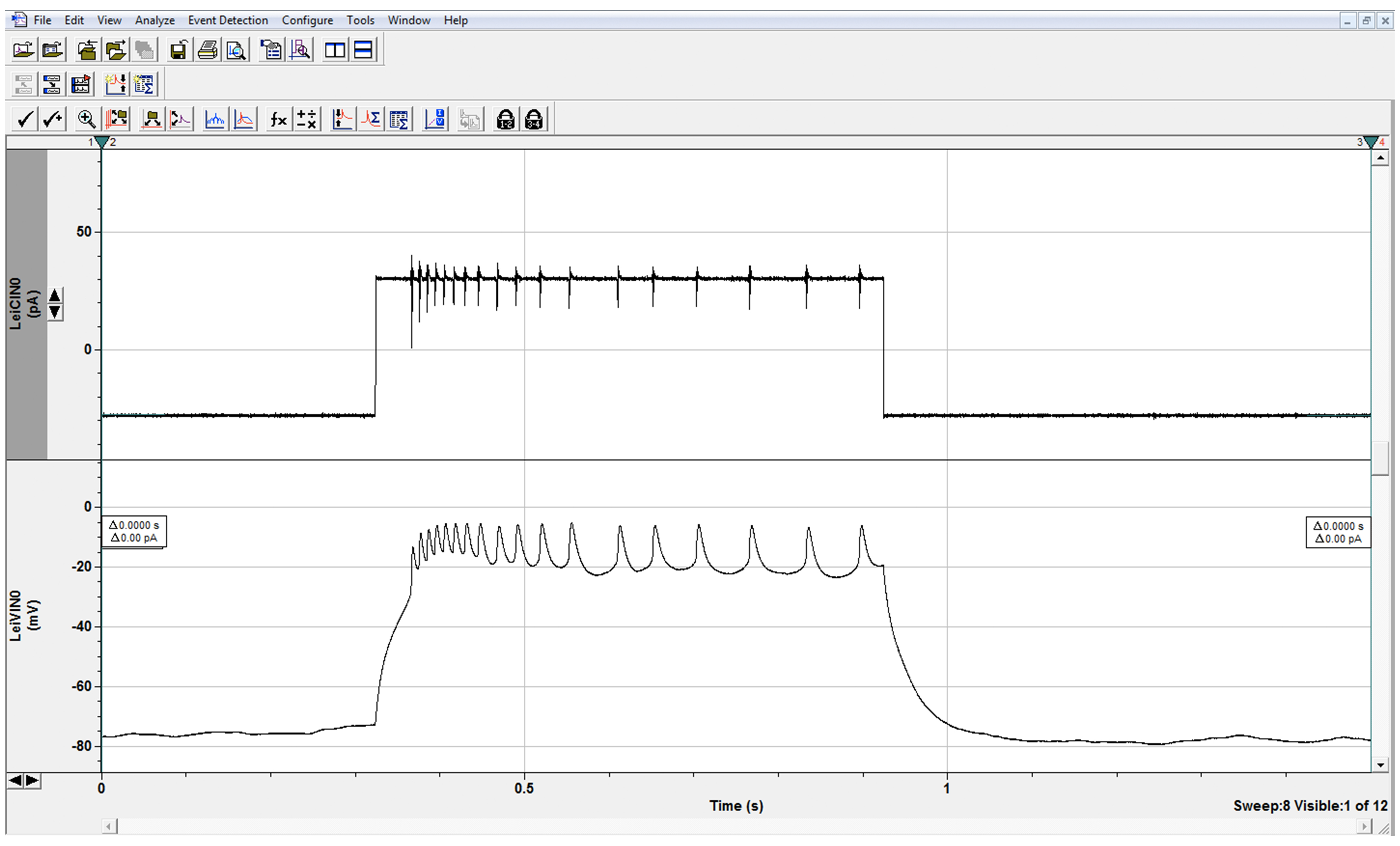
Figure 7. Evoked Firing. Trains of action potential spikelets in LNs in response to depolarizing current step as shown. In current clamp, one can study the evoked firing properties of neurons, including spikelet amplitude, and spikelet frequency as a function of stimulus strength. This wild-type neuron fired 18 spikelets in response to a 30 pA stimulus. Higher or lower spikelet frequency in mutant neurons compared to frequency in control cells indicates hyper- or hypoexcitability, respectively. The amplitude peaks are smaller than those seen in other organisms as we are recording from the inexcitable cell soma and action potentials are generated on the distant processes.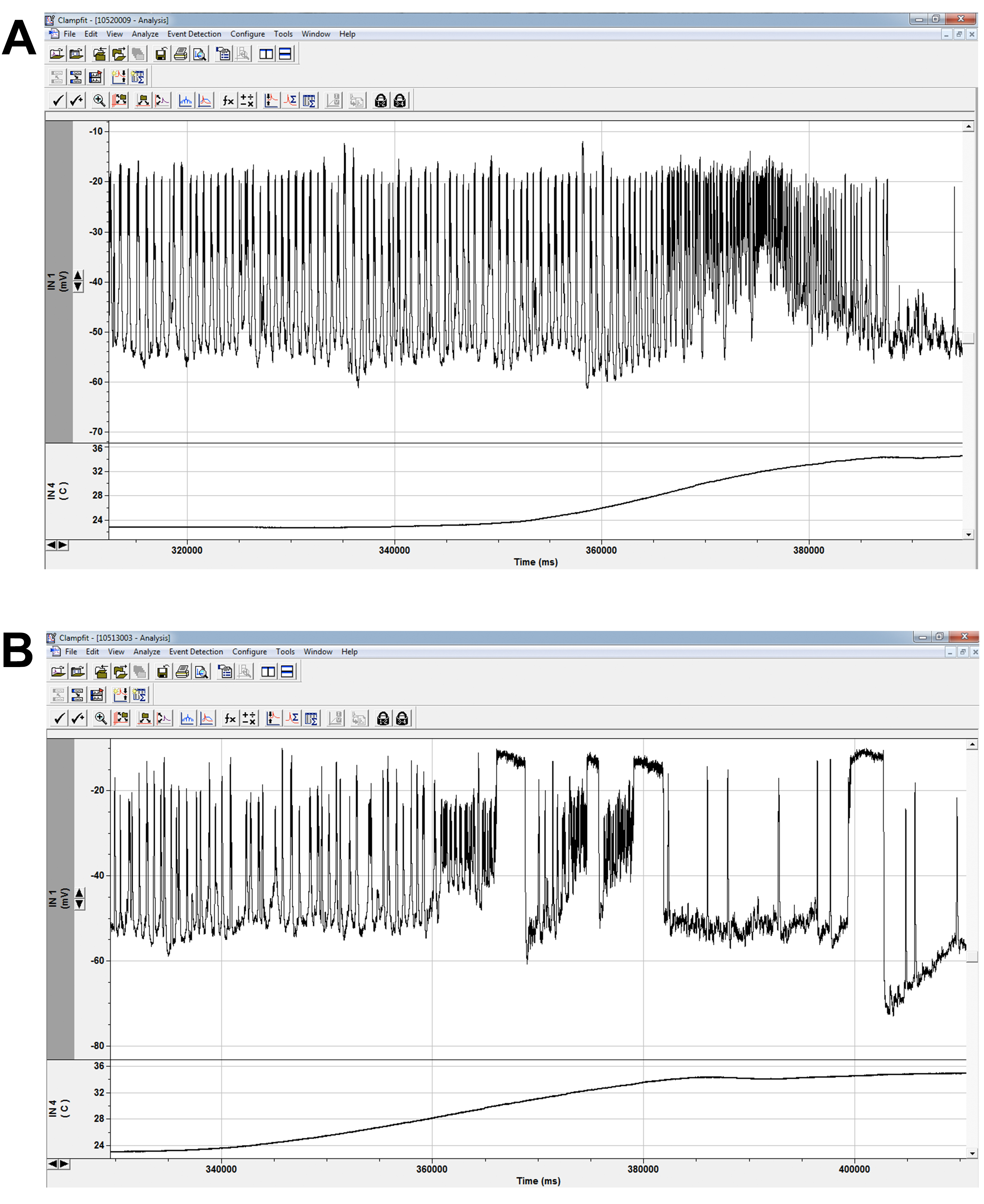
Figure 8. Spontaneous firing and heating protocol. Spontaneous firing recording in control (A) and mutant (B) LNs as temperature is elevated from 22 °C to 35 °C. Due to our ex vivo preparation, we can run any protocol under different conditions. Since our lab studies mutant lines with temperature-sensitive phenotypes, recordings at high temperature are imperative for our understanding of the channel and cellular consequences of channel mutations. Major trace aspects we analyze are spike and burst thresholds, frequencies, and durations. Wildtype cells exhibit increased firing at elevated temperatures (A). The sustained depolarizations in mutants (B) were largely a heat-sensitive phenotype and indicated a gain-of-function mutation for sodium channel activity (Schutte et al., 2016). (Additional equipment: Harvard Apparatus In-Line Solution Heater and Harvard Apparatus Temperature Controller)
Recipes
- Dissecting Solution
External Solution 920 μl (see Recipe 2 below)
L-cysteine stock solution (2 mg/ml in external solution) 80 μl
NaOH (1 M) 5 μl
Papain 9 U
Vortex well after adding papain and wait 20-30 min until the solution is clear to use
It is best to use the dissection solution within 4 h - External Solution for current-clamp and for making dissection solution
Sodium chloride 122 mM
Potassium chloride 3.0 mM
Calcium chloride 1.8 mM
Magnesium chloride 0.8 mM
Glucose 5.0 mM
HEPES 10 mM
(+)-Tubocurarine 20 μM
Picrotoxin 10 μM
When recording spontaneous firing, omit PTX and curarine
Adjust pH 7.2 with NaOH and adjust osmolarity to 250-255 mOsm L-1
External solution can be kept at 4 °C for a week - External Solution for voltage-clamp
Sodium chloride 122 mM
Potassium chloride 3.0 mM
Colbalt chloride 1.8 mM
Magnesium chloride 0.8 mM
Glucose 5.0 mM
HEPES 10 mM
Tetraethylammonium (TEA) 2.5 mM
4-aminopyridine (4-AP) 1.0 mM
(+)-Tubocurarine 20 μM
Picrotoxin 10 μM
Adjust pH 7.2 with NaOH and adjust osmolarity to 250-255 mOsm L-1
External solution can be kept at 4 °C for a week - Internal (Pipette) Solution for current-clamp
Potassium gluconate 102 mM
Sodium chloride 17 mM
Calcium chloride 0.085 mM
Magnesium chloride 1.7 mM
HEPES 8.5 mM
EGTA 0.94 mM
ATP 25 mM (2.5 mg/ml)*
Adjust pH to 7.2 with KOH and adjust osmolarity to 232-235 mOsm L-1
The solution can be aliquoted into 1-2 ml aliquots and store at -20 °C for up to one year
Note: *Just before experiments, defrost one aliquot and add ATP 25 mM (2.5 mg/ml). - Internal (Pipette) Solution for voltage-clamp
Cesium hydroxide 102 mM
D-gluconic acid 102 mM
Sodium chloride 17 mM
Calcium chloride 0.085 mM
Magnesium chloride 1.7 mM
HEPES 8.5 mM
EGTA 0.94 mM
ATP 25 mM (2.5 mg/ml)*
Adjust pH to 7.2 with CsOH and adjust osmolarity to 232-235 mOsm L-1
The solution can be aliquoted into 1-2 ml aliquots and store at -20 °C for up to one year
Note: *Just before experiments, defrost one aliquot and add ATP 25 mM (2.5 mg/ml).
Acknowledgments
This work was generously supported by NIH Grant NS083009 to DKOD and GAANN fellowship P200A120207 to AJR.
Competing interests
The authors declare no conflicts of interest.
References
- Allada, R., White, N. E., So, W. V., Hall, J. C. and Rosbash, M. (1998). A mutant Drosophila homolog of mammalian clock disrupts circadian rhythms and transcription of period and timeless. Cell 93(5): 791-804.
- Bolduc, F. V., Bell, K., Cox, H., Broadie, K. S. and Tully, T. (2008). Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci 11(10): 1143-1145.
- Cervantes-Sandoval, I., Chakraborty, M., MacMullen, C. and Davis, R. L. (2016). Scribble scaffolds a signalosome for active forgetting. Neuron 90(6): 1230-1242.
- Genç, Ö., Dickman, D. K., Ma, W., Tong, A., Fetter, R. D. and Davis, G. W. (2017). MCTP is an ER-resident calcium sensor that stabilizes synaptic transmission and homeostatic plasticity. Elife 6: e22904.
- Gu, H. and O'Dowd, D. K. (2006). Cholinergic synaptic transmission in adult Drosophila Kenyon cells in situ. J Neurosci 26(1): 265-272.
- Gu, H. and O'Dowd, D. K. (2007). Whole cell recordings from brain of adult Drosophila. J Vis Exp(6): 248.
- Guo, F., Yu, J., Jung, H. J., Abruzzi, K. C., Luo, W., Griffith, L. C. and Rosbash, M. (2016). Circadian neuron feedback controls the Drosophila sleep--activity profile. Nature 536(7616): 292-297.
- Guven-Ozkan, T., Busto, G. U., Schutte, S. S., Cervantes-Sandoval, I., O'Dowd, D. K. and Davis, R. L. (2016). MiR-980 is a memory suppressor microRNA that regulates the autism-susceptibility gene A2bp1. Cell Rep 14(7): 1698-1709.
- Hige, T., Aso, Y., Modi, M. N., Rubin, G. M. and Turner, G. C. (2015). Heterosynaptic plasticity underlies aversive olfactory learning in Drosophila. Neuron 88(5): 985-998.
- Liu, S., Sawada, T., Lee, S., Yu, W., Silverio, G., Alapatt, P., Millan, I., Shen, A., Saxton, W., Kanao, T., Takahashi, R., Hattori, N., Imai, Y. and Lu, B. (2012). Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet 8(3): e1002537.
- Nagel, K. I. and Wilson, R. I. (2016). Mechanisms underlying population response dynamics in inhibitory interneurons of the Drosophila antennal lobe. J Neurosci 36(15): 4325-4338.
- Ni, J. D., Baik, L. S., Holmes, T. C. and Montell, C. (2017). A rhodopsin in the brain functions in circadian photoentrainment in Drosophila. Nature 545(7654): 340-344.
- Pallos, J., Bodai, L., Lukacsovich, T., Purcell, J. M., Steffan, J. S., Thompson, L. M. and Marsh, J. L. (2008). Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum Mol Genet 17(23): 3767-3775.
- Parker, L., Padilla, M., Du, Y., Dong, K. and Tanouye, M. A. (2011). Drosophila as a model for epilepsy: bss is a gain-of-function mutation in the para sodium channel gene that leads to seizures. Genetics 187(2): 523-534.
- Qiao, J., Zou, X., Lai, D., Yan, Y., Wang, Q., Li, W., Deng, S., Xu, H. and Gu, H. (2014). Azadirachtin blocks the calcium channel and modulates the cholinergic miniature synaptic current in the central nervous system of Drosophila. Pest Manag Sci 70(7): 1041-1047.
- Schutte, R. J., Schutte, S. S., Algara, J., Barragan, E. V., Gilligan, J., Staber, C., Savva, Y. Q., Smith, M. A., Reenan, R., O’Dowd, D. K. (2014). Knock-in model of Dravet syndrome reveals a constitutive and conditional reduction in sodium current. J Neurophysiol 112: 903-912.
- Schutte, S. S., Schutte, R. J., Barragan, E. V. and O'Dowd, D. K. (2016). Model systems for studying cellular mechanisms of SCN1A-related epilepsy. J Neurophysiol 115(4): 1755-1766.
- Sears, J. C. and Broadie, K. (2017). Fragile X mental retardation protein regulates activity-dependent membrane trafficking and trans-synaptic signaling mediating synaptic remodeling. Front Mol Neurosci 10: 440.
- Sheeba, V., Fogle, K. J., Kaneko, M., Rashid, S., Chou, Y. T., Sharma, V. K. and Holmes, T. C. (2008a). Large ventral lateral neurons modulate arousal and sleep in Drosophila. Curr Biol 18(20): 1537-1545.
- Sheeba, V., Gu, H., Sharma, V. K., O'Dowd, D. K. and Holmes, T. C. (2008b). Circadian- and light-dependent regulation of resting membrane potential and spontaneous action potential firing of Drosophila circadian pacemaker neurons. J Neurophysiol 99(2): 976-988.
- Sun, L., Gilligan. J., Staber, C., Schutte, R. J., Nguyen, V., O’Dowd, D. K., Reenan, R. (2012). A knock-in model of human epilepsy in Drosophila reveals a novel cellular mechanism associated with heat-induced seizure. Journal Neurosci 32: 14145-14155.
- Sun, X., Ran, D., Zhao, X., Huang, Y., Long, S., Liang, F., Guo, W., Nucifora, F. C., Jr., Gu, H., Lu, X., Chen, L., Zeng, J., Ross, C. A. and Pei, Z. (2016). Melatonin attenuates hLRRK2-induced sleep disturbances and synaptic dysfunction in a Drosophila model of Parkinson's disease. Mol Med Rep 13(5): 3936-3944.
- Wilson, R. I., Turner, G. C. and Laurent, G. (2004). Transformation of olfactory representations in the Drosophila antennal lobe. Science 303(5656): 366-370.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Roemmich, A. J., Schutte, S. S. and O'Dowd, D. K. (2018). Ex vivo Whole-cell Recordings in Adult Drosophila Brain. Bio-protocol 8(14): e2467. DOI: 10.21769/BioProtoc.2467.
- Gu, H. and O'Dowd, D. K. (2006). Cholinergic synaptic transmission in adult Drosophila Kenyon cells in situ. J Neurosci 26(1): 265-272.
Category
Neuroscience > Nervous system disorders > Cellular mechanisms
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.