- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation of Nuclei in Tagged Cell Types (INTACT), RNA Extraction and Ribosomal RNA Degradation to Prepare Material for RNA-Seq

(*contributed equally to this work) Published: Vol 8, Iss 7, Apr 5, 2018 DOI: 10.21769/BioProtoc.2458 Views: 16236
Reviewed by: Renate WeizbauerJuliane K IshidaHiroyuki Hirai
Original research article
The authors used this protocol in:

Feb 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Gene expression is dynamically regulated on many levels, including chromatin accessibility and transcription. In order to study these nuclear regulatory events, we describe our method to purify nuclei with Isolation of Nuclei in TAgged Cell Types (INTACT). As nuclear RNA is low in polyadenylated transcripts and conventional pulldown methods would not capture non-polyadenylated pre-mRNA, we also present our method to remove ribosomal RNA from the total nuclear RNA in preparation for nuclear RNA-Seq.
Keywords: Gene expressionBackground
Isolating specific cell types for gene expression experiments reduces the noise and increases the precision of the experiment and the number of differently expressed genes found. Various methods for cell type-specific studies are widely used, each one with their strengths and weaknesses (reviewed in Bailey-Serres, 2013). Isolating specific regulatory compartments, such as nuclei (from organelles, ribosomes, cytosol, etc.) provides further precision in dissecting the molecular events in regulation of gene expression. Here we describe a method that allows isolating nuclei of specific cell types from frozen tissue, suited for experiments where nuclear gene expression is to be studied (e.g., RNA-Seq of nuclear RNA, ATAC-Seq, ChIP-Seq, etc.). Furthermore, we describe the processing of RNA that leads to material suited to be the input for an RNA-Seq experiment. The protocols described here were used with rice root tissue (Oryza sativa cv. Nipponbare) (Reynoso et al., 2018), but they are based on previous protocols developed for Arabidopsis (Deal and Henikoff, 2010 and 2011) and tomato (Ron et al., 2014).
The first part of this protocol, the INTACT method (for Isolation of Nuclei Tagged in specific Cell Types) allows in vivo affinity labeling and subsequent purification of nuclei from a cell type of interest. This is achieved through cell type-specific expression of a tripartite nuclear tagging fusion protein (NTF) consisting of a nuclear envelope targeting domain, GFP, and the biotin ligase recognition peptide (BLRP). Co-expression of NTF along with the E. coli biotin ligase gene codon optimized for rice, BirA, in the cell type of interest results in the production of fluorescently labeled, biotinylated nuclei specifically in that cell type. These labeled nuclei can then be affinity purified from a crude tissue homogenate using streptavidin-coated magnetic beads, thus allowing access to RNA and chromatin from the cell type of interest.
The second part of the method describes the processing of nuclear RNA to produce a sample that is suitable to be the input for RNA-Seq library preparation. Typically for eukaryotic samples, ribosomal RNA (rRNA) and organellar RNA is removed from RNA samples by polyA-pulldown methods. However, nuclear mRNA contains pre-mRNA in many stages of processing, most not polyadenylated, while many of the polyadenylated mRNAs are rapidly exported. Hence, to profile the transcripts at different stages of processing, polyA-isolation methods are not suited. Inspired by previous work (Morlan et al., 2012; Gregory Smaldone, personal communication), we developed a method to remove ribosomal RNA (rRNA) from rice samples, specifically using the INTACT-purified nuclei as our input. The steps of this method include isolation of total RNA from INTACT-purified nuclei, removal of residual genomic DNA, annealing of DNA probes to rRNA, degradation of rRNA with RNase specific to RNA:DNA hybrids, and degradation of the DNA probes. At the end of the protocol, the RNA sample is ready for RNA-Seq library construction.
Part I: INTACT purification of nuclei
Materials and Reagents
- Pipette tips (P20, P200 and P1000, nuclease-free, e.g., USA Scientific, catalog numbers: 1123-1710 , 1120-8780 , 1126-7510 )
- (Optional) 13 ml Falcon tube
- 30 μm cell strainer (Sysmex, CellTrics, catalog number: 04-004-2326 )
- 15 ml Falcon tubes (nuclease-free, e.g., VWR, catalog number: 89039-666 )
- 1.5 ml Eppendorf tubes (nuclease-free, e.g., Denville Scientific, catalog number: C2170 )
- Pasteur pipettes (nuclease-free, e.g., Phenix Research Products, catalog number: PP-137038C )
- Slide
- Coverslip
- PCR tube strips (nuclease-free, e.g., USA Scientific, catalog number: 1402-2708 )
- 0.22 μm syringe filters (e.g., Merck, catalog number: SLGP033RB )
- 50 ml Falcon tubes (nuclease-free, e.g., VWR, catalog number: 89039-658 )
- Aluminum foil
- Transgenic plant tissue with biotin-tagged nuclei
- INTACT binary vectors (Ron et al., 2014; Reynoso et al., 2018)
Note: They will be available at https://gateway.psb.ugent.be/search. These plasmids allow inserting a T-DNA with the promoter of your choice into the plant species of your choice. - Liquid nitrogen
- M-280 Streptavidin Dynabeads (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11205D )
- NaOH
- MOPS (Sigma-Aldrich, catalog number: M1254-25G )
- Sodium chloride (NaCl) (e.g., Fisher Scientific, catalog number: S271 )
- Potassium chloride (KCl) (e.g., Fisher Scientific, catalog number: BP366-1 )
- Ethylenediaminetetraacetic acid (EDTA) (e.g., Fisher Scientific, catalog number: S311 )
- EGTA (Sigma-Aldrich, catalog number: E3889-25G )
- Spermine (Sigma-Aldrich, catalog number: S1141 )
- Spermidine (Sigma-Aldrich, catalog number: S2626 )
- Protease Inhibitor Cocktail (Sigma-Aldrich, catalog number: P9599 ) or cOmplete mini protease inhibitor tablets EDTA-free (Roche Diagnostics, catalog number: 11836170001 )
- Triton X-100 (Sigma-Aldrich, catalog number: 648466 )
- Propidium Iodide (PI) stain (Sigma-Aldrich, catalog number: P4170 )
- 10 N NaOH (see Recipes)
- 0.5 M MOPS, pH 7 (see Recipes)
- 1 M NaCl (see Recipes)
- 2 M KCl (see Recipes)
- 0.5 M EDTA (see Recipes)
- 0.5 M EGTA (see Recipes)
- 0.2 M spermine (see Recipes)
- 0.2 M spermidine (see Recipes)
- 10% Triton X-100 (see Recipes)
- Nuclei purification buffer base (see Recipes)
- NPB and NPBt buffers (see Recipes)
- Propidium Iodide (PI) stains (see Recipes)
Equipment
- Pipettes (P20, P200, P1000; e.g., Eppendorf, catalog numbers: 3123000039 , 3123000055 , 3123000063 )
- Ceramic mortars and pestles (wipe clean with RNaseZap [Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9780 ] or other RNase removal product)
- Centrifuge for 15 ml Falcon tubes (e.g., Eppendorf, model: 5810 R )
- DynamagTM-15 Magnet (Thermo Fisher Scientific, model: DynamagTM-15, catalog number: 12301D )
Note: Can be replaced with a homemade version. For example, a strong rare earth (neodymium) magnet (e.g., 1 x 10 cm bar) can be taped inside a 50 ml Falcon tube and padding can be added to keep 15 ml Falcon tubes in place next to the magnet (see Figure 1).
Figure 1. Homemade magnet for two 15 ml Falcon tubes. Two neodymium magnets are placed inside 50 ml Falcon tubes held in a tube rack. The attraction between the two magnets holds them in place. To prop the 15 ml tubes to correct position against the magnet, paper towel is taped inside the 50 ml tube. - DynamagTM-2 Magnet (Thermo Fisher Scientific, model: DynamagTM-2, catalog number: 12321D ), or a homemade version of this
- Hemocytometer (SCK Films, In-Cyto, catalog number: DHC-N01-2 )
- BD AdamsTM NutatorTM Single-Speed Orbital Mixer (e.g., Fisher Scientific, catalog number: 14-062)
Manufacturer: BD, catalog number: 421105/DEL . - Fridge and/or cold room at 4 °C
- Fluorescent microscope with filters to visualize either DAPI or PI stain (Edmund Optics, catalog numbers: 86-371 , 67-008 )
Procedure
Figure 2 provides the overview of the procedure, including optional check points.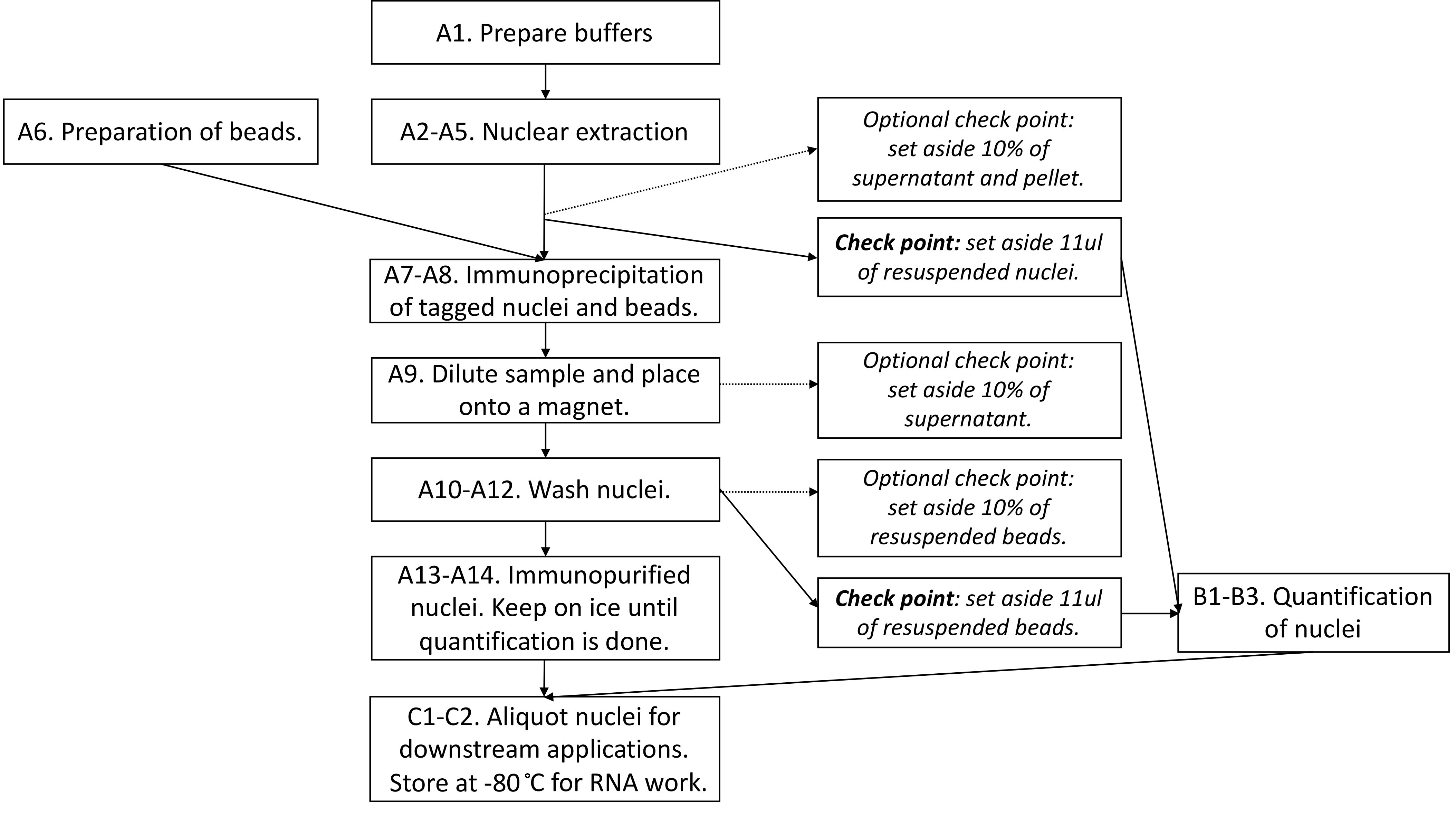
Figure 2. Flowchart summarizing the steps in Part I: INTACT purification of nuclei
Note: For researchers who plan to use INTACT-purified nuclei for Chromatin ImmunoPrecipitation (ChIP) experiments (not described in this protocol): to carry out ChIP experiments, fresh tissue needs to be cross-linked.
- Crosslink proteins to DNA by placing all starting tissue in 30 ml of NPB with 1% formaldehyde (Sigma-Aldrich , F8775) in a 50-ml Falcon tube, and incubating the tissue under vacuum for 10 min.
- Add glycine (Fisher Scientific, BP381) to a final concentration of 0.125 M, place under vacuum for 5 more min.
- Wash tissue 3 times with water and blot dry.
- Isolating nuclei from frozen material
- Prepare fresh NPB and NPBt buffers as needed (~15 ml NPB and 45 ml NBPt per sample) and cool the buffers on ice. We recommend processing maximum eight samples in one go.
- Grind frozen tissue (50-200 1 cm rice root tips. 200 root tips weigh 180 mg) in liquid nitrogen with ceramic mortars and pestles until very fine.
Optional: Pour powder into 13 ml Falcon tubes and weigh the sample. Cool everything with liquid nitrogen and do not let the samples thaw. - Add ~8 ml of ice-cold NPB with a transfer pipette into the mortar and resuspend the powdered tissue by gently grinding it (scale up for larger samples). It is important that NPB does not freeze, so either use minimal amounts of liquid nitrogen for grinding or move to a fresh mortar for this step.
- Filter the resuspension through the 30 μm cell strainer into a 15 ml Falcon tube, and place the Falcon tube on ice while you process rest of your samples.
- Centrifuge the filtered nuclei 1,000 x g for 15 min at 4 °C (2,229 rpm on the Eppendorf 5810 R centrifuge).
- Prepare the streptavidin beads in 1.5 ml tubes (one per sample). Wash 25 μl of Invitrogen M-280 Streptavidin Dynabeads for each sample. To wash the beads, pipette well-mixed beads into a 1.5 ml tube, place on a magnet, remove supernatant, add and mix with 1 ml NPB, place on the magnet, remove supernatant, resuspend in NPB of the original volume (50 μl).
- Move the Falcon tubes from the centrifuge onto ice.
Optional: Mark down the volume of supernatant (SN) and set aside 10% of the supernatant for a Western blot (‘SN1000’–if you want to confirm you have not lost any nuclei/biotin signal in your supernatant).- Discard the supernatant (by careful pouring).
- Resuspend the nuclei in 1 ml of cold NPB by pipetting up and down. In other words, releasing buffer from the pipette on top of the nuclear pellet until the pellet fully resuspends.
- Set aside 11 μl of the resuspended nuclei for quantification and yield calculations (PCR strip works well). See Procedure B.
Optional: Set aside 10% of the resuspension ‘P1000’ (Pellet) for a Western blot (the total amount of biotin signal in the sample).
- Discard the supernatant (by careful pouring).
- Transfer the resuspended nuclei (~1 ml) onto the washed beads (in 1.5 ml tubes). Rotate the tubes on a nutator (at ~20 rpm) at 4 °C for 30 min to bind nuclear envelope biotin onto streptavidin on the beads.
- Dilute the 1 ml of bead-nuclei mixture with 14 ml NPBt in a 15 ml Falcon tube. Mix gently and place on the nutator (at ~20 rpm) at 4 °C for 30 sec. Place the tube on the magnet at 4 °C for 15-20 min.
Optional: Set aside 10% of the supernatant for a Western blot (‘Unbound’). - Carefully remove the supernatant with a Pasteur pipette and gently resuspend the beads in 14 ml NPBt. Mix gently and place on the nutator (at ~20 rpm) at 4 °C for 30 sec. Place the tube on the magnet for 10 min (at 4 °C).
- Repeat Step A10.
- Gently remove the supernatant. Resuspend the beads in 1 ml of NPBt.
Set aside 11 μl of the nuclei for quantification and yield calculations (PCR strip works well). See Procedure B.
Optional: Set aside 10% of the resuspended beads for a Western blot (‘Bound’ to make sure most of your biotinylated signal is here). - Transfer 1 ml resuspension into a 1.5 ml tube and capture on the magnet.
- Remove supernatant, resuspend beads in 20 μl NPB.
Note: Do not freeze nuclei until after they have been counted and aliquoted.
- Prepare fresh NPB and NPBt buffers as needed (~15 ml NPB and 45 ml NBPt per sample) and cool the buffers on ice. We recommend processing maximum eight samples in one go.
- Counting nuclei with a hemocytometer to quantify the yield
- Mix 11 μl nuclei (either on beads from Step A14 or from the resuspended pellet on Step A7c) with 0.5 μl 10 μg/ml Propidium Iodide (PI) stain. Allow nuclei to stain for at least 5 min before imaging.
- Pipette 10 μl of the sample onto a hemocytometer. One hemocytometer has two positions, A and B, for counting nuclei so you need one hemocytometer for counting two samples. The sample is pipetted onto a half-moon shaped area, which pushes the nuclei resuspension between the slide and the coverslip (Figure 3).
- View the samples under fluorescent light and a filter suitable for PI, count the nuclei using the counting grid (Figure 3). Use this to calculate the number of nuclei in total sample.
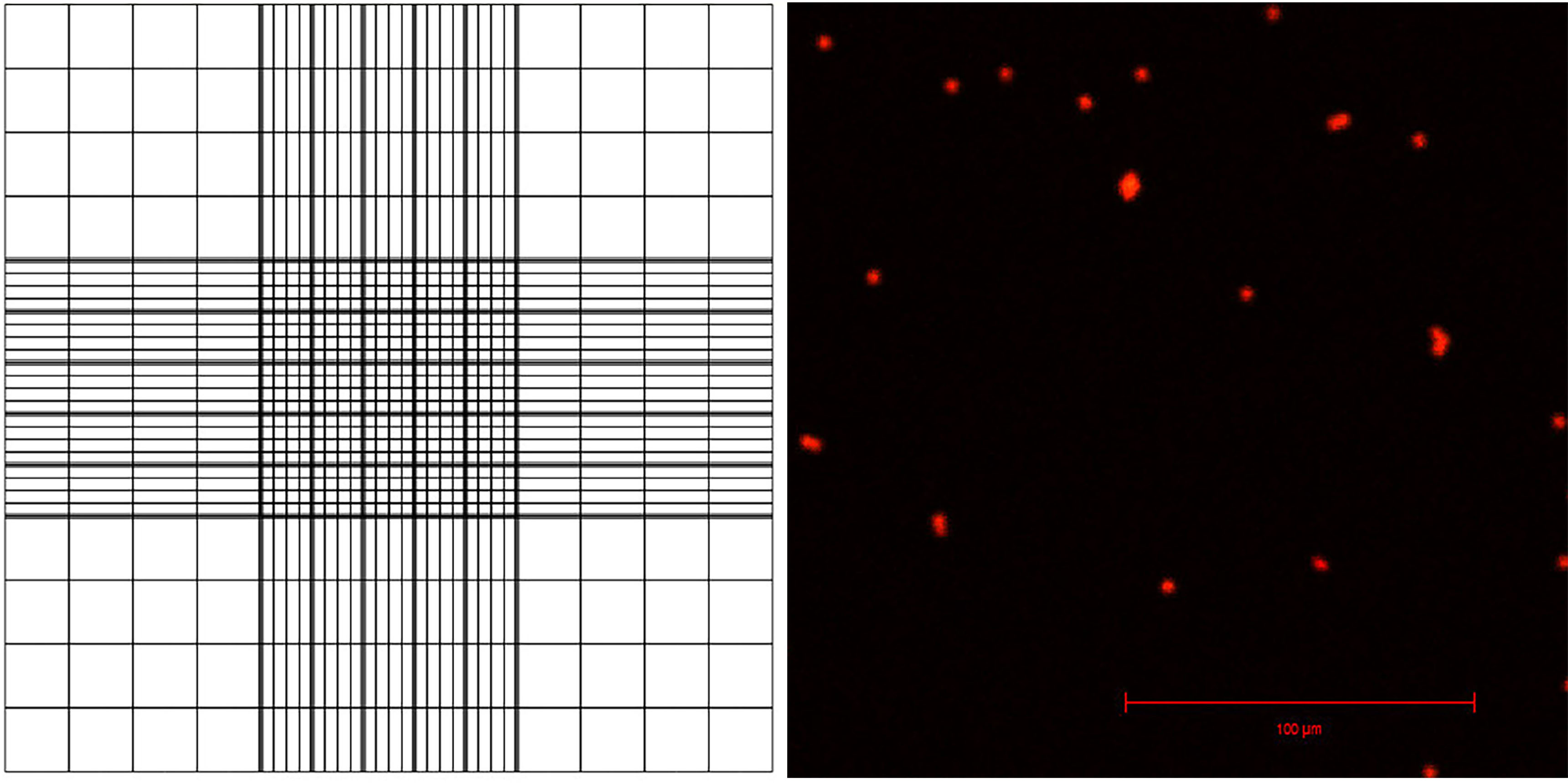
Figure 3. Appearance of the counting grid on a hemacytometer, and visualization of PI-stained nuclei with confocal laser scanning microscopy- For example, you can calculate 16 squares in the corner of the grid, comprising 1/9th of the whole grid (this area is 1 x 1 x 0.1 mm, or 0.1 μl, and represents 1/10,000th of 1 ml).
- Multiply the number of nuclei found inside this area by 10,000 to find out total yield (how many nuclei you have isolated).
Advice:- Typical yields for a strong near-constitutive promoter line (e.g., 35Spro:NTF) from 200 root tips is 150,000-230,000 nuclei.
- Check/count starting number of total nuclei (using nuclei from Step A7c) if you have concerns about your original nuclear isolation and appearance of the nuclei under the microscope.
- Typical yields for a strong near-constitutive promoter line (e.g., 35Spro:NTF) from 200 root tips is 150,000-230,000 nuclei.
- Optional calculations: Background can be calculated by using non-transgenic tissue (‘negative control’) and comparing to constitutive promoter (‘35S-IN’), using both nuclei pulled down with beads and number of total nuclei in the original pellet (P1000):
Efficiency: (35S-IN beads - background)/(35S-IN pellet)
Background: (35S-IN beads) x (negative control beads)/(negative control pellet)
- For example, you can calculate 16 squares in the corner of the grid, comprising 1/9th of the whole grid (this area is 1 x 1 x 0.1 mm, or 0.1 μl, and represents 1/10,000th of 1 ml).
- Mix 11 μl nuclei (either on beads from Step A14 or from the resuspended pellet on Step A7c) with 0.5 μl 10 μg/ml Propidium Iodide (PI) stain. Allow nuclei to stain for at least 5 min before imaging.
- Aliquoting nuclei for RNA and ATAC.
Note: This splitting of the sample is relevant only if you are using the isolated nuclei for an ATAC-seq experiment (described in detail by Bajic et al., 2018) in addition to RNA-Seq.- Calculate how many μl of nuclei you need out of the final 20 μl to get 50,000 nuclei for ATAC. Pipette these onto a PCR strip on ice and start ATAC protocol without freezing (Bajic et al., 2018).
- Use the rest of the nuclei for RNA immediately, or freeze at -80 °C for later RNA work.
- Calculate how many μl of nuclei you need out of the final 20 μl to get 50,000 nuclei for ATAC. Pipette these onto a PCR strip on ice and start ATAC protocol without freezing (Bajic et al., 2018).
Data analysis
The protocol described above needs to be combined with other downstream protocols (RNA-Seq, ATAC-Seq, ChIP-Seq, qRT-PCT) to generate data to be analyzed.
Recipes
- 10 N NaOH
To 80 ml of dH2O, slowly add 40 g of NaOH pellets while continuously stirring - 0.5 M MOPS pH 7
20.93 g MOPS in 200 ml deionized water
Adjust the pH to 7.0 with 10 N NaOH (or NaOH pellets)
Filter sterilize through 0.22 μm filter, aliquot to 40 ml aliquots in 50 ml Falcon tubes, and store at -20 °C - 1 M NaCl
11.688 g NaCl in 200 ml deionized water
Autoclave, store at room temperature - 2 M KCl
29.8205 g KCl in 200 ml deionized water
Autoclave, store at room temperature - 0.5 M EDTA
29.224 g EDTA in 200 ml deionized water
Adjust pH to 8.0 with solid NaOH (> 8 g)
Note: EDTA does not dissolve until the pH has been adjusted.
Autoclave, store at room temperature - 0.5 M EGTA
19.0175 g EGTA in 100 ml deionized water
Adjust pH 8.0 with solid NaOH (~4 g)
Note: EGTA does not dissolve until the pH has been adjusted.
Autoclave, store at room temperature - 0.2 M spermine
1 g in 24.7 ml of deionized water
Filter sterilize through 0.22 μm filter, aliquot, and freeze at -20 °C - 0.2 M spermidine
1 g in 34.4 ml of deionized water.
Filter sterilize through a 0.22 μm filter, aliquot, and freeze at -20 °C - 10% Triton X-100
5 ml Triton X-100
45 ml nuclease-free water
Keep at room temperature - Nuclei Purification Buffer (NPB) base
NPB base can be filter sterilized through a 0.22 μm filter and kept at 4 °C for up to 3 months. Typically 60 ml of NPB base is needed per sample
20 mM MOPS (pH 7)
40 mM NaCl
90 mM KCl
2 mM EDTA
0.5 mM EGTA
For 60 ml (one sample):
2.4 ml 0.5 M MOPS
2.4 ml 1 M NaCl
2.7 ml 2 M KCl
240 μl 0.5 M EDTA pH 8
60 μl 0.5 M EGTA pH 8
For 1 L (~16 samples):
40 ml 0.5 M MOPS
8 ml 5 M NaCl
45 ml 2 M KCl
4 ml 0.5 M EDTA pH 8
1 ml 0.5 M EGTA pH 8 - NPB and NPBt buffers
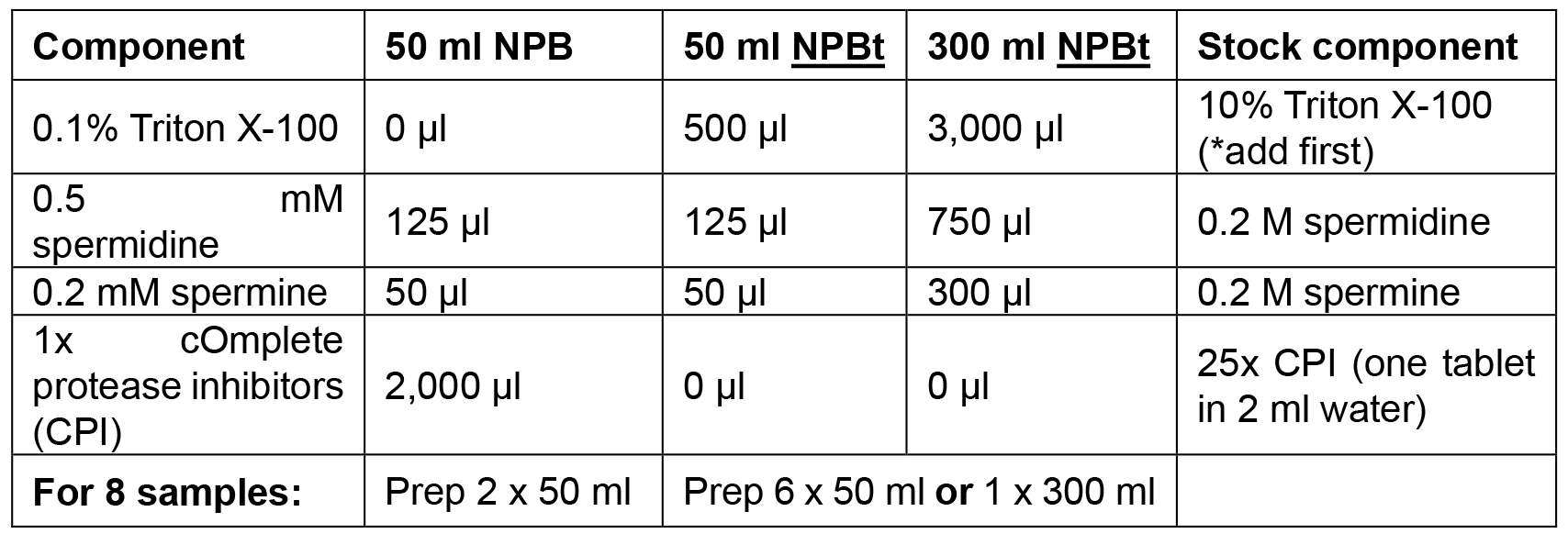
Add Triton X-100, spermidine, spermine and cOmplete protease inhibitors just prior to use and keep on ice or at 4 °C - Propidium Iodide (PI) stains
Prepare 1,000x concentrated stock solution by dissolving 10 mg PI in 1 ml distilled water
Prepare a 1x working solution of 10 μg/ml of PI with diluting the stock 1:1,000 with distilled water
Store solutions at 4 °C, wrapped in aluminum foil to prevent degradation
Part II: Total RNA isolation and rRNA degradation
Materials and Reagents
- Pipette tips (for P10, P20, P200, P1000, nuclease-free e.g., USA Scientific, catalog numbers: 1123-1710 , 1120-8780 , 1126-7510 )
- 1.5 ml Eppendorf tubes (nuclease-free, e.g., Denville Scientific, catalog number: C2170 )
- RNase-free PCR strips (e.g., USA Scientific, catalog number: 1402-2708 )
- 0.22 μm syringe filters (e.g., Merck, catalog number: SLGP033RB )
- Nuclei isolated with INTACT in Part I, or any other suitable starting tissue
- 1 μM stock of DNA probes designed against rRNA sequences
Note: See detailed description on how to design these in Step 1 of the procedure. - RNeasy Micro Kit (QIAGEN, catalog number: 74004 ) or RNeasy Micro Plus Kit (QIAGEN, catalog number: 74034 )
Note: Both kits contain the needed components, but often one is available cheaper than the other. - 70% ethanol
- RNase-free water
- Turbo DNase Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM2238 )
- Agencourt RNAClean XP beads (Beckman Coulter, catalog number: A66514 )
Alternatively: Use AMPureXP beads (Beckman Coulter, catalog number: A63881 ). - Hybridase Thermostable RNase H (Epicentre, catalog number: H39500 )
- Tris base (e.g., Fisher Scientific, catalog number: BP154-1 )
- Hydrochloric acid (HCl, e.g., Sigma-Aldrich, catalog number: 435570 )
- Sodium chloride (NaCl) (e.g., Fisher Scientific, catalog number: S271 )
- Magnesium chloride (MgCl2) (e.g., Sigma-Aldrich, catalog number: M8266 )
- 5x Hybridization buffer H1 (see Recipes)
- 10x Hybridase buffer H2 (see Recipes)
Equipment
- Pipettes (P20, P200, P1000; e.g., Eppendorf, catalog numbers: 3123000039 , 3123000055 , 3123000063 )
- DynamagTM-2 Magnet (Thermo Fisher Scientific, model: DynamagTM-2, catalog number: 12321D ), or a homemade version of this
- Magnetic rack for PCR strips (e.g., Edge BioSystems, catalog number: 57624 )
- Microcentrifuge (e.g., Eppendorf, catalog number: 5424 series )
- PCR machine (e.g., Thermo Fisher Scientific, Applied BiosystemsTM, model: 2720 )
- NanoDrop spectrophotometer (e.g., Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000 , catalog number: ND-2000)
Software
- Sequence analysis software: SnapGene, Geneious, etc.
Procedure
Figure 4 provides the overview of the protocol, including all the possible stop points and temperatures (theoretically you can stop at these points for up to multiple weeks).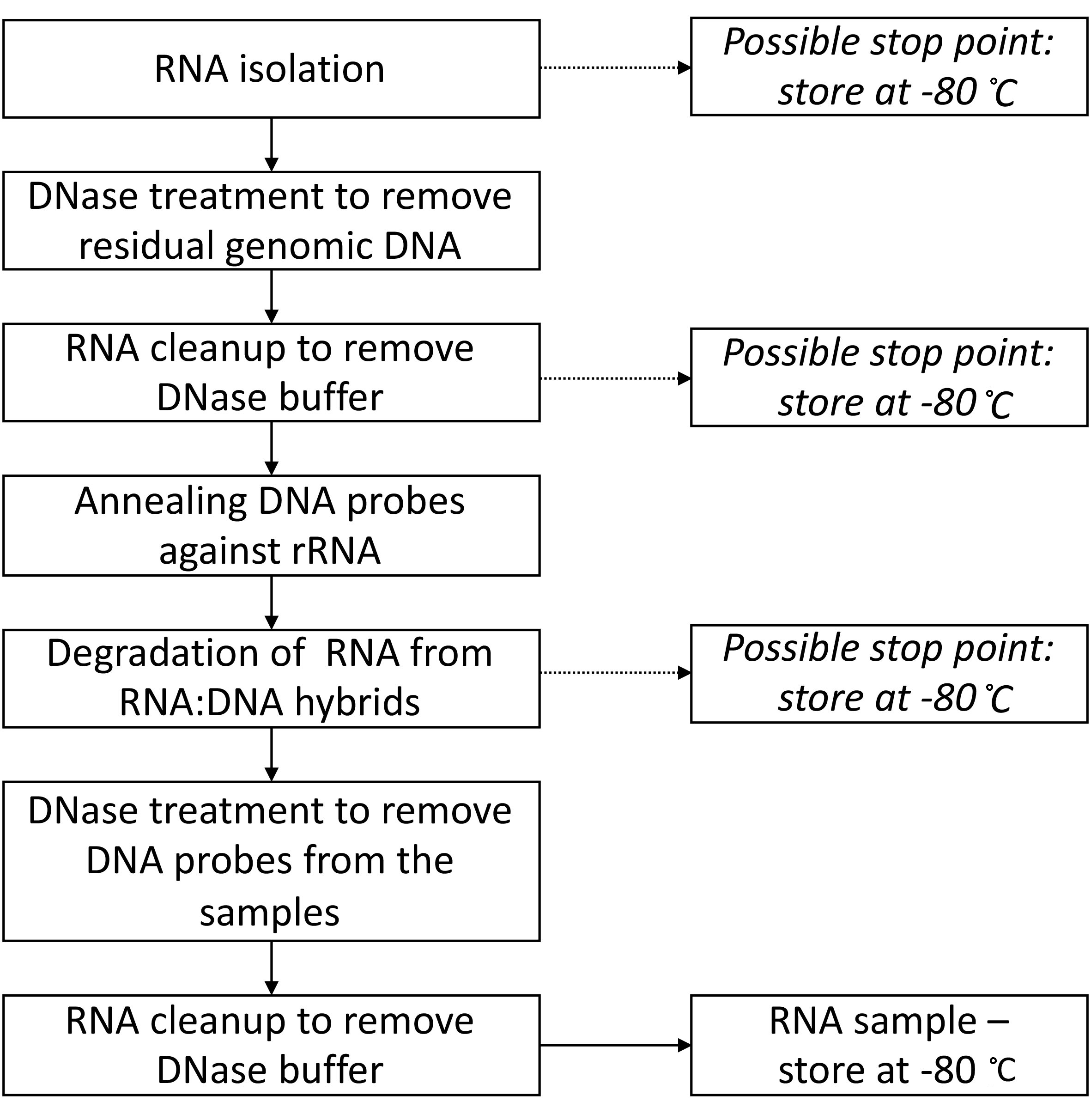
Figure 4. Flowchart summarizing the steps in Part II: Total RNA isolation and rRNA degradation
- Designing DNA probes against rRNA sequences
- Designing the DNA probes against rRNA sequences is a key part of this procedure, as this is how you target RNA for degradation. For targeting nuclear rRNA, you need to obtain the sequences of 5S, 5.8S, 18S and 25S rRNAs and their internal and external transcribed spacers. If you plan to carry out rRNA degradation for total cellular RNA, remember to also target mitochondrial and plastid rRNAs. For many species, these sequences can be found from https://www.ncbi.nlm.nih.gov/nucleotide/.
- Design your probes to be 60 bp long, cover the entire length of rRNA (including the spacer regions as they will be present in nuclear rRNA), and to reverse complement the rRNA sequences. Any sequence analysis software able to convert sequences to reverse complement (SnapGene, Geneious, etc.) can be used.
- Order your probes (DNA oligos) as a pre-made mix at 10 nmol in 1,000 μl of water. This is your 10 μM stock.
- Dilute your stock 10x into a working mix where each probe is at 1 μM.
- Designing the DNA probes against rRNA sequences is a key part of this procedure, as this is how you target RNA for degradation. For targeting nuclear rRNA, you need to obtain the sequences of 5S, 5.8S, 18S and 25S rRNAs and their internal and external transcribed spacers. If you plan to carry out rRNA degradation for total cellular RNA, remember to also target mitochondrial and plastid rRNAs. For many species, these sequences can be found from https://www.ncbi.nlm.nih.gov/nucleotide/.
- Total RNA isolation with QIAGEN RNeasy Micro Kit from nuclei isolated using INTACT
Purify the nuclear RNA using the QIAGEN RNeasy Micro Kit as follows:- Start by adding 350 μl of lysis buffer RLT (with 10 μl beta-mercaptoethanol/1 ml added) to the 20 μl of purified nuclei. Vortex vigorously for 2 min.
- Centrifuge the lysate at 1,000 x g (3,100 rpm) for 2 min at RT to pellet the beads. Place the tube onto a DynamagTM-2 Magnet and transfer the supernatant to a new 1.5 ml tube. Add 350 μl of 70% ethanol and vortex several times to mix.
- Pipette lysate/ethanol mixture into an RNeasy MinElute spin column resting in a 2 ml collection tube and centrifuge at 10,000 x g (9,700 rpm) for 1 min at RT. Discard flowthrough.
- Add 350 μl of buffer RW1 to the column. Centrifuge at 10,000 x g (9,700 rpm) for 1 min at RT. Discard flowthrough and move the column to a new 2 ml collection tube.
- Add 500 μl of buffer RPE to the column. Centrifuge at 10,000 x g (9,700 rpm) for 1 min at RT. Discard flowthrough.
- Add 500 μl of 80% ethanol to the column. Centrifuge at 10,000 x g (9,700 rpm) for 1 min at RT. Discard flowthrough and move the column to a new 2 ml collection tube.
- Open the column lid and centrifuge at top speed (16,000 x g) for 5 min at RT. Discard the flowthrough and place the column into a new 1.5 ml tube.
- Add 20 μl RNase-free water onto the column membrane and allow to stand for 1 min. Centrifuge at 16,000 x g for 1 min at RT.
- Store RNA at -80 °C.
- Start by adding 350 μl of lysis buffer RLT (with 10 μl beta-mercaptoethanol/1 ml added) to the 20 μl of purified nuclei. Vortex vigorously for 2 min.
- DNase I treatment to eliminate genomic DNA contamination
Use DNase I protocol for Turbo DNase I as follows:- Add to 20 μl of RNA:
2 μl of 10x DNase I reaction buffer
1 μl DNase I - Incubate for 30 min at 37 °C.
- Add 2 μl DNase I inactivation reagent (vortex well before adding) and incubate for 5 min at RT (vortex every 1 min).
- Spin down (2,000 x g for 5 min) and recover 20 μl into a new tube.
- Add to 20 μl of RNA:
- RNA cleanup using Agencourt RNAClean XP bead to remove DNase buffer
- Add 1.8 volume (e.g., 36 μl for 20 μl of RNA) of RNAClean XP beads to the reaction.
- Incubate at RT for 10 min.
- Onto a magnet for 5 min.
- Remove most of the supernatant (leave 5 μl behind to avoid pipetting up beads).
- Leave tubes on the magnet, add 200 μl 70% ethanol, let stand for 30 sec.
- Remove all the supernatant.
- Repeat 70% wash as above once more (two washes total).
- Remove as much of the EtOH as possible.
- Air dry beads on the magnet for 10 min (until appear dry, see Figure 5).
- Add 15 μl RNase-free water to the first aliquot dried bead and mix.
- If you have aliquoted samples, pool back together.
- Incubate at RT for 5 min.
- Onto the magnet for 5 min.
- Recover 15 μl eluate.

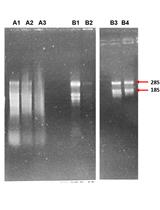
Figure 5. Appearance of Agencourt XP beads. A. After ethanol has been removed, the beads appear glistening and wet. B. Beads ready for elution look dry, the pellet is thinner and often lighter in color.
- Add 1.8 volume (e.g., 36 μl for 20 μl of RNA) of RNAClean XP beads to the reaction.
- NanoDrop for [RNA] and 260/280 so that you know the RNA concentration for adjusting probe concentration.
- rRNA probe hybridization to bind DNA probes to rRNA
- Reaction size for Steps 6 and 7 needs to be 6 μl + 4 μl (total 10 μl) for the buffers to be optimal for each step. You can prepare multiple aliquots of the reaction and pool back before DNase I treatment (Step 8). Successful RNA-Seq libraries have been made from a single 0.1 μg reaction.
- Add following together:
3.8 μl RNA (if this would be more than 1 μg RNA, make up with RNase-free water)
1.2 μl 5x Hybridization buffer H1 (Recipe 1)
1.0 μl probe mix–choose your concentration as follows:- If your RNA amount is 1 μg, use 1 µM/oligo working stock of probe mix.
- If your RNA amount is 0.1 μg, dilute probes to 0.1 µM/oligo.
Note: This is the most common probe concentration in our experiments. - If your RNA amount is 0.01 μg, dilute probes to 0.01 µM/oligo.
- If your RNA amount is under NanoDrop detection range, use 0.01 µM/oligo.
- If your RNA amount is 1 μg, use 1 µM/oligo working stock of probe mix.
- Incubate as follows in a PCR machine:
95 °C for 2 min
Ramp down to 45 °C at 0.1 °C/sec
45 °C for 5 min
Hold at 45 °C
- Reaction size for Steps 6 and 7 needs to be 6 μl + 4 μl (total 10 μl) for the buffers to be optimal for each step. You can prepare multiple aliquots of the reaction and pool back before DNase I treatment (Step 8). Successful RNA-Seq libraries have been made from a single 0.1 μg reaction.
- Hybridase® (thermostable RNase H) reaction to digest RNA from RNA:DNA hybrids
- Prepare a master mix that you preheat to 45 °C (in a hot block):
1 μl 10x Hybridase buffer H2 (Recipe 2)
1 μl Hybridase (5 U/μl)
2 μl nuclease-free water - Add 4 μl of the MM to hybridization reaction still at 45 °C (keep in the PCR machine).
- Incubate at 45 °C for 30 min.
- Remove to ice.
- Prepare a master mix that you preheat to 45 °C (in a hot block):
- DNase I treatment to digest DNA oligos/probes from RNA pool
- Use DNase I protocol for Turbo DNase I.
- Add to 40 μl of RNA (for four aliquots pooled):
4 μl of 10x DNase I reaction buffer
2 μl DNase I - Incubate for 30 min at 37 °C.
- Add 2 μl DNase I inactivation reagent (vortex well before adding) and incubate for 5 min at RT (vortex every 1 min). The minimum volume of inactivation reagent to add is 2 μl–so even smaller volumes (e.g., 10 μl), add 2 μl.
- Spin down (2,000 x g for 5 min) and recover 43 μl into a new tube.
- Use DNase I protocol for Turbo DNase I.
- RNA cleanup using Agencourt RNAClean XP beads
- Add 1.8 volume (e.g., 77.4 μl for 43 μl of RNA) of RNAClean XP beads to rxn.
- Incubate at RT for 10 min.
- Onto a magnet for 5 min.
- Remove most of the supernatant (leave 5 μl behind to avoid pulling up beads).
- Leave tubes on the magnet, add 200 μl 70% ethanol, let stand for 30 sec.
- Remove all the supernatant.
- Repeat 70% wash as above once more (two washes total).
- Remove as much of the EtOH as possible.
- Air dry beads on the magnet for 10 min (until appear dry, see Figure 5).
- Add 10 μl RNase-free water to the first aliquot dried bead and mix.
- If you have aliquoted samples, pool back together.
- Incubate at RT for 5 min.
- Onto the magnet for 5 min.
- Recover 10 μl eluate. Store RNA at -80 °C.
- Add 1.8 volume (e.g., 77.4 μl for 43 μl of RNA) of RNAClean XP beads to rxn.
Data analysis
The protocol described above needs to be combined with other downstream protocols (RNA-Seq) to generate data to be analyzed.
Recipes
- 5x Hybridization buffer H1 (RNase-free)
0.5 M Tris
1 M NaCl
Adjust pH to 7.0 with HCl
Filter sterilize through a 0.22 μm filter, aliquot to 1 ml aliquots, and store at -20 °C - 10x Hybridase buffer H2 (RNase-free)
500 mM Tris
1 M NaCl
200 mM MgCl2
Adjust pH to 7.4 with HCl
Filter sterilize through a 0.22 μm filter, aliquot to 1 ml aliquots, and store at -20 °C
Acknowledgments
This work was supported by the United States National Science Foundation (NSF) Plant Genome grant No. IOS-1238243 to R.B.D., S.M.B., N.S. and J.B.-S. and by a Finnish Cultural Foundation postdoctoral fellowship (to K.K.). An NSF Research Experiences for Undergraduates supplement (IOS-1238243) and site program (DBI-1461297) supported S.C and J.V., respectively.
Authors declare no conflicts of interest or competing interests.
References
- Bajic, M., Maher, K. A. and Deal, R. B. (2018). Identification of open chromatin regions in plant genomes using ATAC-Seq. Methods Mol Biol 1675: 183-201.
- Bailey-Serres, J. (2013). Microgenomics: Genome-scale, cell-specific monitoring of multiple gene regulation tiers. Annu Rev Plant Bio 64: 293-325.
- Deal, R. B. and Henikoff, S. (2010). A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell 18(6): 1030-1040.
- Deal, R. B. and Henikoff, S. (2011). The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat Protoc 6(1): 56-68.
- Morlan, J. D., Qu, K. and Sinicropi, D. V. (2012). Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS One 7(8): e42882.
- Reynoso, M.A, Pauluzzi, G., Kajala, K., Cabanlit, S., Velasco, J., Bazin, J., Deal, R.B., Sinha, N.R., Brady, S.M., Bailey-Serres J. (2018). Nuclear transcriptomes at high resolution using retooled INTACT. Plant Physiol 176(1): 270-281.
- Ron, M., Kajala, K., Pauluzzi, G., Wang, D., Reynoso, M. A., Zumstein, K., Garcha, J., Winte, S., Masson, H., Inagaki, S., Federici, F., Sinha, N., Deal, R. B., Bailey-Serres, J. and Brady, S. M. (2014). Hairy root transformation using Agrobacterium rhizogenes as a tool for exploring cell type-specific gene expression and function using tomato as a model. Plant Physiol 166(2): 455-469.
- Townsley, B.T., Covington, M.F., Ichihashi, Y., Zumstein, K., Sinha, N.R. (2015). BrAD-seq: Breath Adapter Directional sequencing: a streamlined, ultra-simple and fast library preparation protocol for strand specific mRNA library construction. Front Plant Sci 6: 366.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Reynoso, M. A., Pauluzzi, G. C., Cabanlit, S., Velasco, J., Bazin, J., Deal, R., Brady, S., Sinha, N., Bailey-Serres, J. and Kajala, K. (2018). Isolation of Nuclei in Tagged Cell Types (INTACT), RNA Extraction and Ribosomal RNA Degradation to Prepare Material for RNA-Seq. Bio-protocol 8(7): e2458. DOI: 10.21769/BioProtoc.2458.
- Reynoso, M.A, Pauluzzi, G., Kajala, K., Cabanlit, S., Velasco, J., Bazin, J., Deal, R.B., Sinha, N.R., Brady, S.M., Bailey-Serres J. (2018). Nuclear transcriptomes at high resolution using retooled INTACT. Plant Physiol 176(1): 270-281.
Category
Plant Science > Plant molecular biology > RNA > RNA sequencing
Plant Science > Plant molecular biology > RNA > RNA extraction
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.