- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Improving CRISPR Gene Editing Efficiency by Proximal dCas9 Targeting
.jpg)
Published: Vol 7, Iss 15, Aug 5, 2017 DOI: 10.21769/BioProtoc.2432 Views: 10930
Reviewed by: Jihyun KimVera Karolina SchoftAnonymous reviewer(s)
Original research article
The authors used this protocol in:

3-Apr 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) systems function as an adaptive immune system in bacteria and archaea for defense against invading viruses and plasmids (Barrangou and Marraffini, 2014). The effector nucleases from some class 2 CRISPR-Cas systems have been repurposed for heterologous targeting in eukaryotic cells (Jinek et al., 2012; Cong et al., 2013; Mali et al., 2013; Zetsche et al., 2015). However, the genomic environments of eukaryotes are distinctively different from that of prokaryotes in which CRISPR-Cas systems have evolved. Mammalian heterochromatin was found to be a barrier to target DNA access by Streptococcus pyogenes Cas9 (SpCas9), and nucleosomes, the basic units of the chromatin, were also found to impede target DNA access and cleavage by SpCas9 in vitro (Knight et al., 2015; Hinz et al., 2015; Horlbeck et al., 2016; Isaac et al., 2016). Moreover, many CRISPR-Cas systems characterized to date often exhibit inactivity in mammalian cells and are thus precluded from gene editing applications even though they are active in bacteria or on purified DNA substrates. Thus, there is a need to devise a means to alleviate chromatin inhibition to increase gene editing efficiency, especially on difficult-to-access genomic sites, and to enable use of otherwise inactive CRISPR-Cas nucleases for gene editing need. Here we describe a proxy-CRISPR protocol for restoring nuclease activity of various class 2 CRISPR-Cas nucleases on otherwise inaccessible genomic sites in human cells via proximal targeting of a catalytically dead Cas9 (Chen et al., 2017). This protocol is exemplified here by using Campylobacter jejuni Cas9 (CjCas9) as nuclease and catalytically dead SpCas9 (SpdCas9) as proximal DNA binding protein to enable CjCas9 to cleave the target for gene editing using single stranded DNA oligo templates.
Keywords: CRISPR-Cas nucleaseBackground
By creating targeted chromosomal DNA double strand breaks (DSBs) or single strand breaks (nicks) or serving as a DNA binding module for other DNA modification effectors, programmable endonucleases have become an important tool for genome modification in eukaryotic cells (Gaj et al., 2013). In response to targeted DNA breaks, host cells can invoke various repair pathways to mend the damages to maintain the genome integrity. Insertions and/or deletions derived from NHEJ repair errors can be capitalized for gene knockout and homologous recombination can be exploited for introducing pre-determined changes on gene of interest by providing a DNA donor. In addition to these more traditional gene editing applications, catalytically inactive forms of programmable endonucleases are increasingly used as DNA binding modules for other DNA modification effectors, such as cytidine deamination enzymes (Komor et al., 2016). However, no matter which forms of programmable nucleases are utilized, target site binding is the prerequisite step and local chromatin structure can determine whether or not or how efficiently a programmable nuclease can bind the target site (Knight et al., 2015; Hinz et al., 2015; Horlbeck et al., 2016; Isaac et al., 2016, Chen et al., 2017). We hypothesize that binding at proximal locations by a programmable DNA binding protein could change the local chromatin structure and render an otherwise inaccessible target site accessible for binding.
Previous generations of programmable nucleases, such as meganucleases, zinc finger nucleases (ZFNs), and transcription activator-like effector nucleases (TALENs), solely rely on protein structure to recognize target sites, and thus re-targeting of these nucleases requires rather laborious protein structural change. In contrast, class 2 CRISPR-Cas effector nucleases use protein structure to recognize a protospacer adjacent motif (PAM) and employ CRISPR RNA (crRNA) to bind the target site adjacent to the PAM. Because PAM is typically a short DNA sequence, such as 5’-NGG-3’ for SpCas9, and thus occurs frequently in a genome, re-targeting of CRISPR-Cas nucleases is a simple process of changing the crRNA sequence by molecular cloning or chemical synthesis. This targeting modality makes CRISPR-Cas systems very suitable for use as nucleases or as DNA binding proteins. This protocol combines these two utilities together to expand CRISPR gene editing capability. The CRISPR-Cas system used as nuclease must be orthogonal to the CRISPR-Cas system used as DNA binding protein to avoid binding site sharing. In general, different subtypes of class 2 CRISPR-Cas systems (e.g., type II-A, type II-B, type II-C, and type V) are orthogonal to one another. Within each subtype, some systems are highly divergent and could be also orthogonal to one another, but they need to be experimentally verified. Currently, it is highly recommended to use SpdCas9 as proximal DNA binding protein, for SpCas9 is the most robust system in mammalian cells to date, although it can also be inactive at certain genomic sites. However, it is anticipated that more robust Cas9 systems will be developed for use as DNA binding proteins.
Materials and Reagents
- Pipette tips (Thermo Fisher Scientific, Thermo ScientificTM, catalog numbers: 2140 , 2149 , 2065E , 2069 , and 2079E )
- 75 cm2 U-shaped canted neck cell culture flask with vent cap (Corning, catalog number: 430641U )
- Microcentrifuge tubes, 1.5 ml (Sigma-Aldrich, catalog number: T6649 )
- Tissue culture plate, 6-well (Corning, Costar®, catalog number: 3516 )
- Serological pipettes
5 ml (Corning, Costar®, catalog number: 4051 )
10 ml (Corning, Costar®, catalog number: 4488 )
25 ml (Corning, Costar®, catalog number: 4489 )
- Cuvettes
- Coverslip
- 50-ml conical centrifuge tube (Corning, catalog number: 430828 )
- Human K562 cells (ATCC, catalog number: CCL-243 )
- Catalytically inactive Streptococcus pyogenes Cas9 (SpdCas9) plasmid DNA (from MilliporeSigma; see Figure 1A)
- SpCas9 sgRNA plasmid constructs with the guide sequences 5’-CCAAGGGTGAGGCCGGGAAG-3’ and 5’-CATCTCCCCCATGTACACCT-3’ for binding two human POR target sites (from MilliporeSigma; see Figure 1B)
- Campylobacter jejuni Cas9 (CjCas9) plasmid DNA (from MilliporeSigma; see Figure 1C)
- CjCas9 sgRNA plasmid construct with the guide sequence 5’-TTCGCCAGTACGAGCTTGTG-3’ for binding a human POR target site (from MilliporeSigma; see Figure 1D)
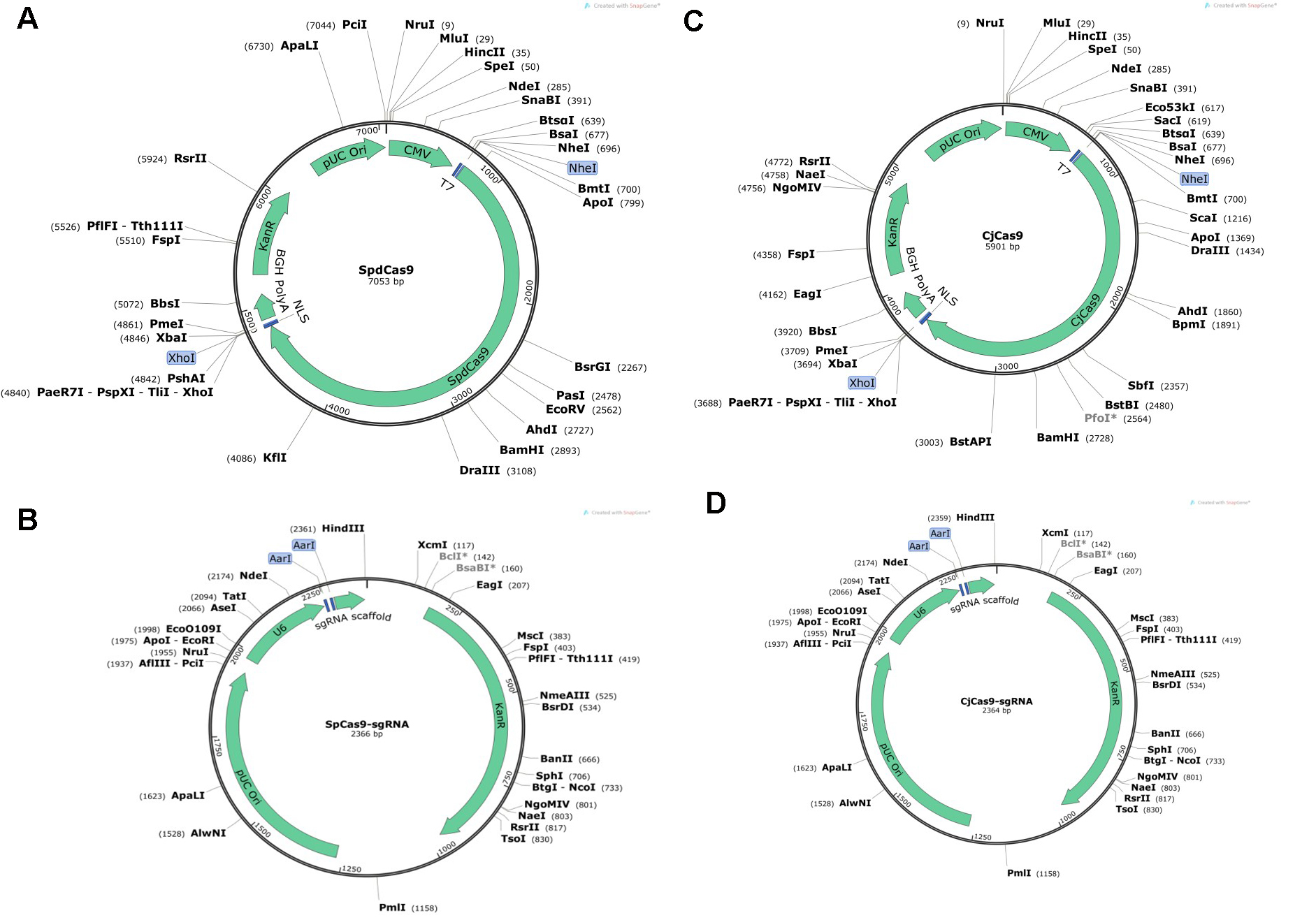
Figure 1. Vector maps
- Single stranded DNA oligo donor for introducing a diagnostic EcoRI site at the CjCas9 cleavage site in POR. The oligo sequence is: 5’-CACCCTTGGTCTCCCCTTTCCAGCATTCGCCAGTACGAGCGAATTCTTGTGGTCCACACCGACATAGATGCGGCCAAGGTGTACATGG-3’ (Underlined: EcoRI site)
Note: Synthesize the oligo at 0.2 µmol scale and purify by PAGE. Re-suspend the oligo in 10 mM Tris buffer (pH 7.6) at 200 µM.
- PCR primers for amplification of the targeted POR genomic region: forward 5’-CTCCCCTGCTTCTTGTCGTAT-3’, reverse 5’-ACAGGTCGTGGACACTCACA-3’
- Alcohol
- Hank’s balanced salt solution (Sigma-Aldrich, catalog number: H6648 )
- Nucleofector instrument (Lonza, catalog number: AAB-1001 )
- Amaxa Cell Line Nucleofector Kit V (Lonza, catalog number: VCA-1003 )
- GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich, catalog number: NA2010 )
- JumpStart Taq ReadyMix (Sigma-Aldrich, catalog number: P2893 )
- GenElute PCR Clean-Up Kit (Sigma-Aldrich, catalog number: NA1020 )
- EcoRI restriction enzyme (New England Biolabs, catalog number: R0101S )
- 10% Mini-PROTEAN TGX Precast Protein Gels, 15-well, 15 µl (Bio-Rad Laboratories, catalog number: 4561036 )
- Tris buffered saline (Sigma-Aldrich, catalog number: T5912 )
- Gel loading buffer (Sigma-Aldrich, catalog number: G2526 )
- DirectLoad Wide Range DNA Marker (Sigma-Aldrich, catalog number: D7058 )
- Iscove’s modified Dulbecco’s medium (Sigma-Aldrich, catalog number: I3390 )
- Fetal bovine serum (FBS) (Sigma-Aldrich, catalog number: F2442 )
- L-glutamine, 200 mM (Sigma-Aldrich, catalog number: G7513 )
- K562 culture medium (see Recipes)
Equipment
- Water bath (Polyscience)
- Laminar flow hood for sterile tissue culture, biosafety level 2 approved (Thermo Fisher Scientific)
- Microbiological incubator with 37 °C, atmospheric CO2 (Thermo Fisher Scientific)
- Pipettes (Gilson)
- Bench-top centrifuge (Eppendorf, model: 5417 C )
- Mini-PROTEAN Tetra Vertical Electrophoresis Cell (Bio-Rad Laboratories, catalog number: 1658004 )
- PCR thermocycler (Bio-Rad Laboratories, catalog number: 1861096 )
Software
- ImageJ (https://imagej.nih.gov/ij/)
Procedure
- Target site selection and oligo design for sgRNA expression
- Nuclease cleavage site selection will be dependent on gene editing objective. For gene knockout, it is recommended to select cleavage sites in the first two coding exons or in protein functional domains. For gene correction or homologous recombination (HR), the cleavage site should be as close as possible to the nucleotide to be changed or to the target integration site to maximize the efficiency. For cytidine deamination-based editing, the nicking site will be dependent on the cytidine deamination window.
- Select a proximal dCas9 binding site upstream and downstream of the cleavage site. The dCas9 binding sites each must be separated from the nuclease binding site by about 10 bp to avoid potential steric hindrance, but beyond the 10 bp separation the dCas9 binding sites also should be kept as close as possible to the nuclease binding site to maximize the effects. The dCas9 binding sites can be on either strand of the DNA.
- One of the advantages of CRISPR-Cas effector nucleases over previous generations of programmable nucleases as genome editing tools is the simplicity of nuclease retargeting via straightforward oligo design and sgRNA expression plasmid cloning once target sites are selected. In this illustrative experiment, the two SpdCas9 proximal binding sites are: 5’-CCAAGGGTGAGGCCGGGAAGCGG-3’, and 5’-CAAGGTGTACATGGGGGAGATGG-3’; and the CjCas9 binding site is: 5’-TTCGCCAGTACGAGCTTGTGGTCCACAC-3’; where the underlined nucleotides are protospacer adjacent motifs (PAMs). Therefore, the DNA oligo sequences for sgRNA expression plasmid cloning are: forward 5’-ACCGCCAAGGGTGAGGCCGGGAAG-3’ and reverse 5’-AAACCTTCCCGGCCTCACCCTTGG-3’ for SpdCas9 binding site 1; forward 5’-ACCGCAAGGTGTACATGGGGGAGA-3’ and reverse 5’-AAACTCTCCCCCATGTACACCTTG-3’ for SpdCas9 binding site 2; and forward 5’-ACCGTTCGCCAGTACGAGCTTGTG-3’ and reverse 5’-AAACCACAAGCTCGTACTGGCGAA-3’ for the CjCas9 binding site; where the underlined nucleotides in the forward sequences are a constant overhang for ligation to a human U6 promoter and the underlined nucleotides in the reverse sequences are a constant overhang for ligation to a SpdCas9 sgRNA scaffold or a CjCas9 sgRNA scaffold in cloning vectors.
- Nuclease cleavage site selection will be dependent on gene editing objective. For gene knockout, it is recommended to select cleavage sites in the first two coding exons or in protein functional domains. For gene correction or homologous recombination (HR), the cleavage site should be as close as possible to the nucleotide to be changed or to the target integration site to maximize the efficiency. For cytidine deamination-based editing, the nicking site will be dependent on the cytidine deamination window.
- Transfection and gene editing analysis
- Culture K562 cells in Iscove’s modified Dulbecco’s medium, supplemented with 10% FBS and 2 mM L-glutamine. Maintain the culture at 37 °C and 5% CO2. Split the culture every 2-3 days or when the culture reaches 1-2 million cells per ml by transferring 1/10 or 1/5 of the culture to a new culture flask containing fresh medium pre-warmed to 37 °C to make a 1:10 or a 1:5 split (see Note 1).
- Split the culture to 0.25 million cells per ml one day before transfection. The culture will reach about 0.5 million cells per ml for transfection the next day (see Note 1).
- Prepare three plasmid DNA solutions for transfection in 1.5-ml microcentrifuge tubes (see Note 2):
- 5 µg of SpdCas9 plasmid DNA, 4.2 µg of CjCas9 plasmid DNA, 3 µg each of the two SpdCas9 sgRNA plasmid constructs and the CjCas9 sgRNA plasmid construct, and 1.5 µl of 200 µM of single stranded DNA oligo donor.
- 4.2 µg of CjCas9 plasmid DNA, 3 µg of the CjCas9 sgRNA plasmid construct, and 1.5 µl of 200 µM of single stranded DNA oligo donor as no proximal dCas9 targeting control.
- 1.5 µl of 200 µM of single stranded DNA oligo donor as oligo-only control. Keep the tubes on ice before transfection.
- Add 2 ml of fresh culture medium to each well of a 6-well plate and equilibrate the medium at 37 °C and 5% CO2 for at least 20 min before transfection.
- Set the Nucleofector program to T-016 (see Note 3). Unseal the cuvettes and disposable transfer pipettes and place them inside a cell culture hood.
- Count the cells with a glass hemocytometer:
- Clean the hemocytometer and coverslip with alcohol before use.
- Gently swirl the flask to make sure the cells are evenly distributed. Take 10 ul of cells and add to the chamber under the coverslip. Cells will be drawn out by capillary action and spread evenly on the hemocytometer.
- Count the cells with 10x objective under a microscope. Using a hand tally counter, count the cells in all four corner squares with 16 smaller squares. Only count the cells within a square or on the right-hand or bottom boundary line.
- Average the total number by four and then multiply by 10,000 (104). This will be the number of cells per ml in the culture.
- Transfer the required amount of cells to a sterile 50-ml conical centrifuge tube. Each transfection reaction will require 1.0 million cells (see Note 4). Centrifuge the cells at 200 x g at room temperature for 5 min. Carefully remove the culture medium by aspiration.
- Resuspend the cell pellet in 20 ml of Hank’s balanced salt solution. Centrifuge at 200 x g at room temperature for 5 min and carefully remove the wash fluid by aspiration. Gently re-suspend the cell pellet in nucleofection solution V to 1.0 million cells per 100 µl (see Note 5).
- Pipette 100 µl of the cells into a 1.5-ml tube containing a plasmid DNA solution and mix thoroughly but gently by pipetting up and down 3-5 times. Transfer the whole content to a nucleofection cuvette and perform the transfection immediately using the T-016 program.
- Add a pipette full of pre-equilibrated culture medium to the transfected cells inside the cuvette with a disposable transfer pipette and immediately transfer the whole content into a well containing pre-equilibrated culture medium. Culture the cells at 37 °C and 5% CO2 immediately after nucleofection.
- Extract genomic DNA 3 days after transfection using GenElute Mammalian Genomic DNA Miniprep Kit. PCR amplify the POR genomic region using JumpStart Taq ReadyMix with the forward primer 5’-CTCCCCTGCTTCTTGTCGTAT-3’ and the reverse primer 5’-ACAGGTCGTGGACACTCACA-3’ and the following cycling condition: 98 °C for 2 min for initial denaturation; 34 cycles of 98 °C for 15 sec, 62 °C for 30 sec, and 72 °C for 45 sec; and a final extension at 72 °C for 5 min.
- Purify PCR products with GenElute PCR Clean-Up Kit. Digest purified PCR products (about 500 ng per digestion) with EcoRI (20 U) at 37 °C for 2 h. Resolve digestion products on a pre-cast 10% acrylamide gel from Bio-Rad Laboratories using DirectLoad Wide Range DNA Marker as markers. Figure 2 illustrates an example of expected results.
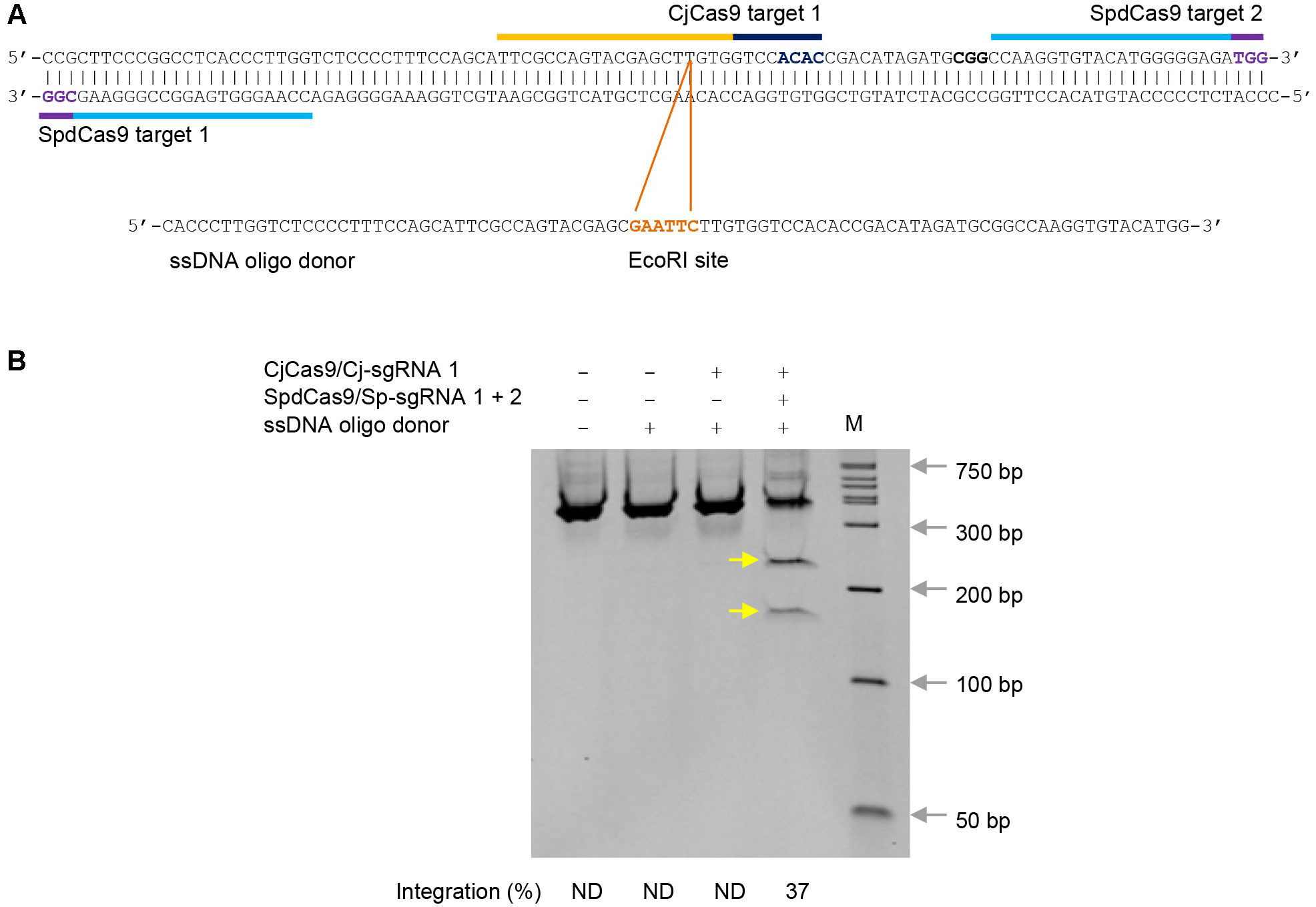
Figure 2. ssDNA oligo templated gene editing in K562 cells by CjCas9 using proximal dCas9 targeting. A. Campylobacter jejuni Cas9 (CjCas9) and catalytically inactive Streptococcus pyogenes Cas9 (SpdCas9) target sites in the human POR locus and an ssDNA oligo donor carrying a diagnostic EcoRI site. Targets are indicated by bars and PAMs are highlighted in dark blue (CjCas9) and purple (SpdCas9). B. EcoRI digestion analysis of ssDNA oligo templated gene editing. EcoRI site integration efficiency (%) was determined by ImageJ. The two EcoRI restriction fragments are indicated by yellow arrows. The sgRNA numbers correspond to the target numbers in (A). ND, not determined. M, wide-range DNA markers.
- Culture K562 cells in Iscove’s modified Dulbecco’s medium, supplemented with 10% FBS and 2 mM L-glutamine. Maintain the culture at 37 °C and 5% CO2. Split the culture every 2-3 days or when the culture reaches 1-2 million cells per ml by transferring 1/10 or 1/5 of the culture to a new culture flask containing fresh medium pre-warmed to 37 °C to make a 1:10 or a 1:5 split (see Note 1).
Data analysis
Use ImageJ to measure the band intensity for the uncut band and EcoRI digested bands. Sequence integration efficiency (%) is determined by the formula: 100 x (b + c)/(a + b + c), where a is the integrated intensity of the undigested PCR product, and b and c are the integrated intensities of each restriction digestion product.
Notes
- When working with adherent cells, split the culture every 2-3 days or when the culture reaches approximately 80% confluency (approximately 80% of the surface is occupied by cells). Split the culture two days before transfection. Seed the culture at about 20% confluency so that the culture will be at about 60-80% confluency at the time of cell preparation for transfection.
- Prepare plasmid DNA at a concentration ≥ 2 µg/µl to limit the total volume to ≤ 10 µl for each transfection.
- Different cell lines require different nucleofection programs. Follow the instructions from the manufacturer when working with other cell lines.
- Up to 2 million K562 cells can be used per nucleofection, but it may reduce the transfection efficiency slightly.
- Do not leave cells in the nucleofection solution for a prolonged time (30 min).
Recipes
- K562 culture medium
500 ml Iscove’s modified Dulbecco’s medium
50 ml fetal bovine serum
5 ml 200 mM L-glutamine
Acknowledgments
The authors wish to thank Patrick Sullivan for R&D administrative support and the financial support from MilliporeSigma, a business of Merck KGaA, Darmstadt, Germany. This protocol was originally published as part of Chen et al. (2017)
References
- Barrangou, R. and Marraffini, L. A. (2014). CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol Cell 54(2): 234-244.
- Chen, F., Ding, X., Mei, Y., Seebeck, T., Jiang, Y. and Davis, G. (2017). Targeted activation of diverse CRISPR-Cas systems for mammalian genome editing via proximal CRISPR targeting. Nat Commun 8(14958).
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Gaj, T., Gersbach, C. A. and Barbas, C. F., 3rd (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7): 397-405.
- Hinz, J. M., Laughery, M. F. and Wyrick, J. J. (2015). Nucleosomes inhibit Cas9 endonuclease activity in vitro. Biochemistry 54(48): 7063-7066.
- Horlbeck, M. A., Witkowsky, L. B., Guglielmi, B., Replogle, J. M., Gilbert, L. A., Villalta, J. E., Torigoe, S. E., Tjian, R. and Weissman, J. S. (2016). Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife 5.
- Isaac, R. S. Jiang, F., Doudna, J. A., Lim, W. A., Narlikar, G. J. and Almeida, R. (2016). Nulceosome breathing and remodeling constrain CRISPR-Cas9 function. eLife 5.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Knight, S. C., Xie, L., Deng, W., Guglielmi, B., Witkowsky, L. B., Bosanac, L., Zhang, E. T., El Beheiry, M., Masson, J. B., Dahan, M., Liu, Z., Doudna, J. A. and Tjian, R. (2015). Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science 350(6262): 823-826.
- Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533(7603): 420-424.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., Volz, S. E., Joung, J., van der Oost, J., Regev, A., Koonin, E. V. and Zhang, F. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163(3): 759-771.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chen, F., Ding, X., Feng, Y., Seebeck, T., Jiang, Y. and Davis, G. D. (2017). Improving CRISPR Gene Editing Efficiency by Proximal dCas9 Targeting. Bio-protocol 7(15): e2432. DOI: 10.21769/BioProtoc.2432.
Category
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.