- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

DNA-free Genome Editing of Chlamydomonas reinhardtii Using CRISPR and Subsequent Mutant Analysis

(*contributed equally to this work) Published: Vol 7, Iss 11, Jun 5, 2017 DOI: 10.21769/BioProtoc.2352 Views: 16051
Reviewed by: Vinay PanwarKaisa KajalaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
We successfully introduced targeted knock-out of gene of interest in Chlamydomonas reinhardtii by using DNA-free CRISPR. In this protocol, the detailed procedures of an entire workflow cover from the initial target selection of CRISPR to the mutant analysis using next generation sequencing (NGS) technology. Furthermore, we introduce a web-based set of tools, named CRISPR RGEN tools (http://www.rgenome.net/), which provides all required tools from CRISPR target design to NGS data analysis.
Keywords: Genome editingBackground
We recently reported (Baek et al., 2016) a one-step transformation of the model green microalga Chlamydomonas reinhardtii (Harris, 2001) using preassembled Cas9 protein-guide RNA ribonucleoproteins (RNPs). The manner of DNA-free CRISPR-Cas9 delivery has several advantages such as no need for codon optimization and specific promoters in different species of microalgae. Furthermore, it reduces off-target effects and may also be less cytotoxic in cells because the Cas9 protein is transiently active and then degraded by endogenous proteases in cells (Kim et al., 2014). In addition, the resulting gene-edited microalgae could be exempt from genetically modified organism (GMO) regulations due to the absence of foreign DNA sequences. In this protocol, the detailed procedures of an entire workflow are contained from the initial target selection of CRISPR to the mutant analysis using NGS technology (Bae et al., 2014a and 2014b; Park et al., 2015 and 2017).
Materials and Reagents
- Cas9 protein purification
- 1.5 ml Eppendorf tubes
- Sterile 50 ml conical tubes
- 10 ml syringe
- 0.45 μm syringe filter
- Protein concentrator–100K MWCO (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 88533 )
- Poly-Prep Chromatography columns (Bio-Rad Laboratories, catalog number: 7311550 )
- BL21-Pro cells
- pET28 plasmid containing 6xHis-SpCas9 (Addgene)
- Kanamycin
- Lysozyme
- PMSF
- Ni-NTA agarose beads (QIAGEN, catalog number: 30210 )
- Bradford reagent (Bio-Rad Laboratories, catalog number: 5000205 )
- Bovine serum albumin (BSA)
- Sodium chloride (NaCl)
- Tryptone
- Yeast extract
- IPTG
- Sodium phosphate monobasic (NaH2PO4)
- Imidazole
- Sodium hydroxide (NaOH)
- HEPES
- Ethylenediaminetetraacetic acid (EDTA)
- DL-dithiothreitol (DTT)
- Sucrose
- Glycerol
- LB medium (see Recipes)
- 1 M IPTG stock (see Recipes)
- Lysis buffer (see Recipes)
- Wash buffer (see Recipes)
- Elution buffer (see Recipes)
- Cas9 storage buffer (see Recipes)
- 1.5 ml Eppendorf tubes
- In vitro transcription and library PCR
- 1.5 ml Eppendorf tubes
- Forward and reverse oligos (see Tables 1 and 2)
Table 1. Pre-index primersPre-index forward primer 5’-ACACTCTTTCCCTACACGACGCTCTTCCGATCT gDNA target-3’ Pre-index reverse primer 5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT gDNA target-3’
Table 2. Index primer sequencesindex sequence [i5] Index forward primer sequence D501 tatagcct AATGATACGGCGACCACCGAGATCTACACtatagcctACACTCTTTCCCTACACGAC D502 atagaggc AATGATACGGCGACCACCGAGATCTACACatagaggcACACTCTTTCCCTACACGAC D503 cctatcct AATGATACGGCGACCACCGAGATCTACACcctatcctACACTCTTTCCCTACACGAC D504 ggctctga AATGATACGGCGACCACCGAGATCTACACggctctgaACACTCTTTCCCTACACGAC D505 aggcgaag AATGATACGGCGACCACCGAGATCTACACaggcgaagACACTCTTTCCCTACACGAC D506 taatctta AATGATACGGCGACCACCGAGATCTACACtaatcttaACACTCTTTCCCTACACGAC D507 caggacgt AATGATACGGCGACCACCGAGATCTACACcaggacgtACACTCTTTCCCTACACGAC D508 gtactgac AATGATACGGCGACCACCGAGATCTACACgtactgacACACTCTTTCCCTACACGAC index sequence [i7] Index reverse primer sequence D701 cgagtaat CAAGCAGAAGACGGCATACGAGATcgagtaatGTGACTGGAGTTCAGACGTGT D702 tctccgga CAAGCAGAAGACGGCATACGAGATtctccggaGTGACTGGAGTTCAGACGTGT D703 aatgagcg CAAGCAGAAGACGGCATACGAGATaatgagcgGTGACTGGAGTTCAGACGTGT D704 ggaatctc CAAGCAGAAGACGGCATACGAGATggaatctcGTGACTGGAGTTCAGACGTGT D705 ttctgaat CAAGCAGAAGACGGCATACGAGATttctgaatGTGACTGGAGTTCAGACGTGT D706 acgaattc CAAGCAGAAGACGGCATACGAGATacgaattcGTGACTGGAGTTCAGACGTGT D707 agcttcag CAAGCAGAAGACGGCATACGAGATagcttcagGTGACTGGAGTTCAGACGTGT D708 gcgcatta CAAGCAGAAGACGGCATACGAGATgcgcattaGTGACTGGAGTTCAGACGTGT D709 catagccg CAAGCAGAAGACGGCATACGAGATcatagccgGTGACTGGAGTTCAGACGTGT D710 ttcgcgga CAAGCAGAAGACGGCATACGAGATttcgcggaGTGACTGGAGTTCAGACGTGT D711 gcgcgaga CAAGCAGAAGACGGCATACGAGATgcgcgagaGTGACTGGAGTTCAGACGTGT D712 ctatcgct CAAGCAGAAGACGGCATACGAGATctatcgctGTGACTGGAGTTCAGACGTGT - dNTP mix
- Phusion DNA polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: F530L )
- PCR purification kit
- ATP, CTP, GTP, UTP, 100 mM MgCl2, DEPC-treated water
- T7 RNA polymerase (New England Biolabs, catalog number: M0251L )
- DNase I (RNase-free) (New England Biolabs, catalog number: M0303L )
- RNase inhibitor murine (New England Biolabs, catalog number: M0314L )
- RNeasy MinElute Cleanup Kit (QIAGEN, catalog number: 74204 )
- Illumina Miseq Reagent Kit (v2)
- 1.5 ml Eppendorf tubes
- Transfection
- 1.5 ml Eppendorf tubes
- Sterile 15 ml and 50 ml conical tubes
- Purified Cas9 protein and sgRNA
- Chlamydomonas reinhardtii strains CC-4349 cw15 mt-(Chlamydomonas Resource Center)
- PCR DNA purification kit (MG Med, catalog number: MD008 )
- TAP medium (Harris, 1989 or Thermo Fisher Scientific, GibcoTM, catalog number: A1379801 )
- TAP sucrose solution (see Recipes)
- TAP agar plates (see Recipes)
- Top agar (see Recipes)
- 1.5 ml Eppendorf tubes
Equipment
- Cas9 protein purification
- In vitro transcription and library PCR
- Incubator (JS Research, model: JSGI-050T )
- Thermal cycler (Bio-Rad Laboratories, model: C1000 TouchTM Thermal Cycler )
- Micro centrifuge (LaboGene, model: 1730R )
- Spectrophotometer
- Incubator (JS Research, model: JSGI-050T )
- Transfection
- 100 ml flask
- Spectrophotometer (GE Healthcare, Amersham Biosciences, model: Ultrospec 2100 pro )
- Hemocytometer (Marienfeld-Superior, catalog number : 0650030 )
- Microscope (Olympus, model: CH30 )
- Micro high speed centrifuge (Hanil, model: MICRO 17TR )
- Orbital shaker (N-biotek, model: NB-101M )
- Clean bench (BioFree, model: BF-150BSC )
- Electroporation device (Bio-Rad Laboratories, model: Gene Pulser XcellTM Electroporation Systems )
- Electroporation cuvettes (Bio-Rad Laboratories, 0.4 cm gap)
- 100 ml flask
Procedure
- CRISPR target selection for creating gene knock-outs of interest
- Obtain the coding sequence (CDS) of the gene of interest from an appropriate database. We use Phytozome from JGI (https://phytozome.jgi.doe.gov/pz/portal.html#) (Figure 1).
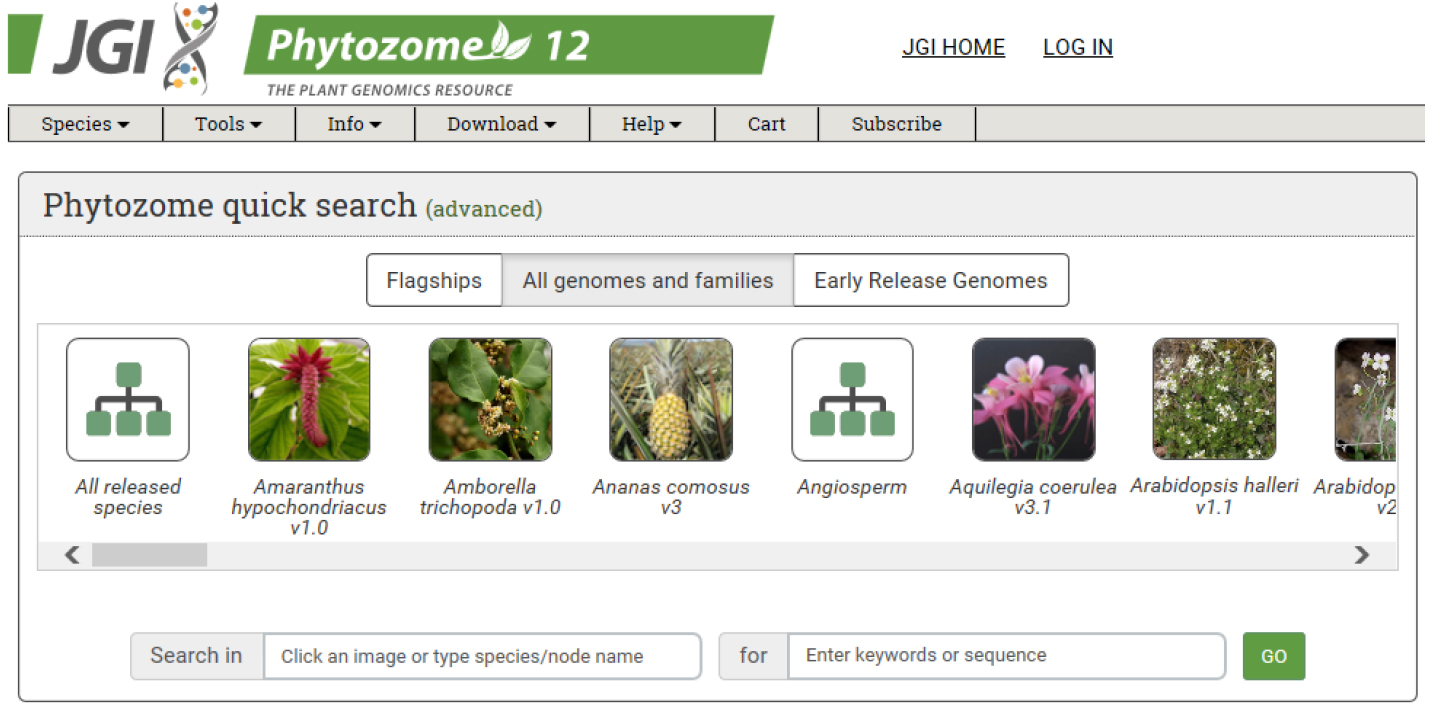
Figure 1. Main page of Phytozome: The plant genome database. The coding sequence information of each gene can be obtained from Phytozome database. - For target design, we use CRISPR RGEN tools (http://www.rgenome.net/), a web-based set of tools for CRISPR target design and next-generation sequencing (NGS) data analysis (Figure 2).
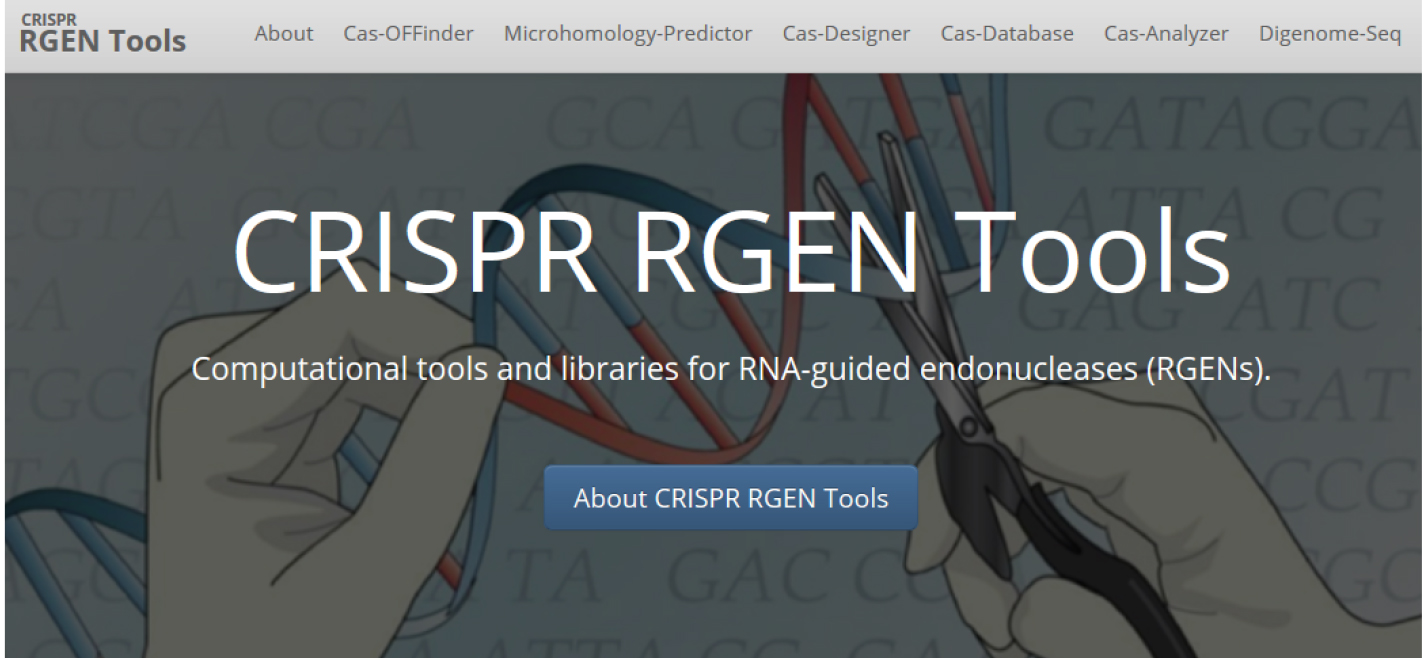
Figure 2. Main page of CRISPR RGEN Tools. A web-based set of tools which provides all required tools from CRISPR target design to NGS data analysis. - In Cas-Designer, select the type of CRISPR endonuclease, such as SpCas9 from Streptococcus pyogenes, and the target genome, in this case that of the model organism Chlamydomonas reinhardtii. Cas-Designer now contains the versions 4 and 5 of it at ‘Plants’ organism type on our website. Then insert the CDS of the gene of interest in the ‘Target Sequence’ box. We recommend choosing target sites within the first half of the full CDS to increase the probability of generating complete knock-out mutations (Figure 3).
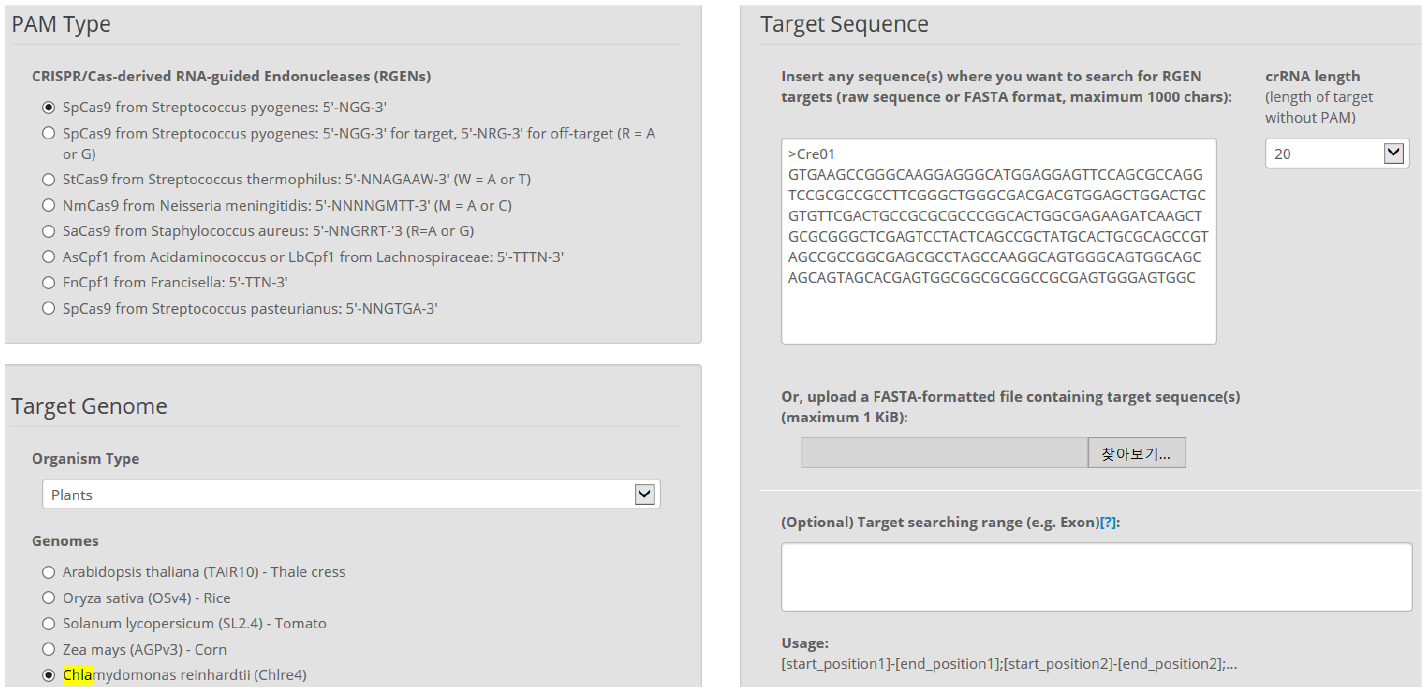
Figure 3. Overview of Cas-Designer. Users can select various types of CRISPR endonucleases and the target genome including Chlamydomonas reinhardtii (versions 4 or 5). - Choose several targets among the candidates from Cas-Designer. You should avoid targets that are associated with potential off-target sites bearing 1-2 mismatches compared to the on-target. Targets with GC content ranging from 30 to 70% and higher out-of-frame scores are recommended (Figure 4).
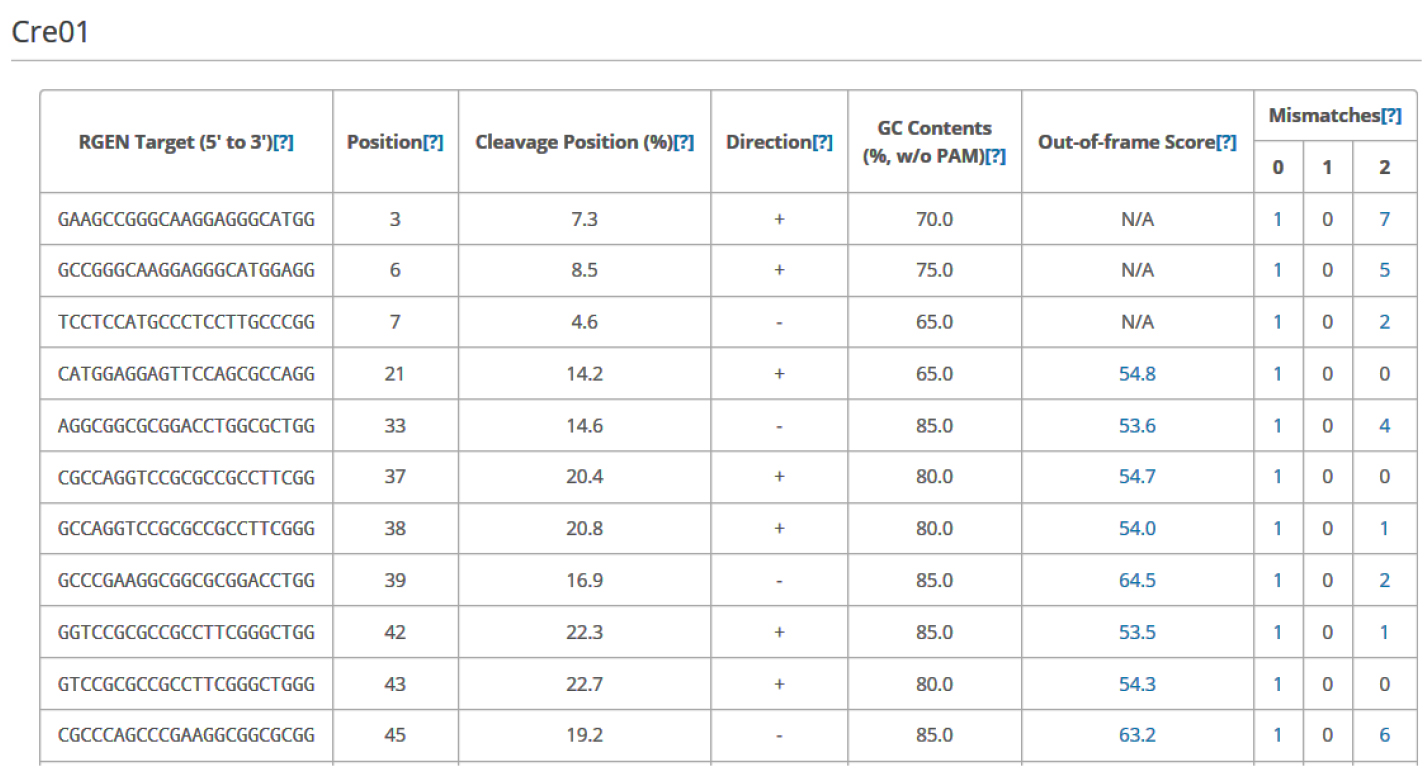
Figure 4. An example data of an output table from Cas-Designer. Cas-Designer shows possible CRISPR target sites from input sequences along with useful information such as GC content, out-of-frame score, and off-target information.
Note: N/A means ‘not applicable’ due to the lack of flanking DNA nucleotides.
- Obtain the coding sequence (CDS) of the gene of interest from an appropriate database. We use Phytozome from JGI (https://phytozome.jgi.doe.gov/pz/portal.html#) (Figure 1).
- Purification of recombinant Cas9 protein
- Transform 50 μl competent BL21-Pro cells with a pET28 vector containing 6xHis-SpCas9.
- Spread the cells on an agar plate containing kanamycin. Incubate the plate overnight at 37 °C.
- Inoculate 10 ml LB medium containing 50 μg/ml kanamycin in a conical tube with a single colony from the plate. Grow to saturation overnight at 37 °C with shaking at 200 rpm.
- The next morning, inoculate 200 ml of LB medium containing kanamycin in a flask with 2 ml saturated overnight culture.
- Grow the cells at 37 °C with shaking at 200 rpm to mid-log phase (to an OD600 of approximately 0.4-0.5).
- Add 160 μl of 1 M IPTG (0.8 mM final concentration) and incubate the cells overnight at 18 °C with shaking at 200 rpm.
- Transfer the culture to a 50 ml conical centrifuge tube and pellet the cells by centrifuging for 20 min at 3,000 x g and 4 °C. Discard the supernatant and repeat the harvest in the same tube.
- Re-suspend the cell pellets in 10 ml lysis buffer (see Recipes).
- Add 20 mg lysozyme and 100 μl of 100 mM PMSF (1 mM final concentration).
- Incubate 1 h on ice.
- Sonicate the cells in a conical tube on ice (amplitude 25%, 1 sec ON/1 sec OFF, pulse 30 times). Repeat the sonication process 5 times.
- Transfer the sonicated cells to a 1.5 ml microtube using micropipette and pellet debris by centrifuging at 4 °C, 11,000 x g for 30 min.
- Filter the supernatant through a 0.45-μm syringe filter and collect it in a 50 ml conical tube.
- Add 2 ml Ni-NTA agarose to the lysate. Swirl agarose beads before adding. Incubate for 1 h with rotation at 4 °C.
- Transfer the mixture to a chromatography column with pipette. Collect the flow-through in a conical tube.
- Wash the column with 10 ml wash buffer (see Recipes). Collect this first batch of wash buffer in a conical tube.
- Repeat the wash. Collect the second batch of wash buffer in a conical tube.
- Elute the bound protein with 10 ml elution buffer (see Recipes). Collect the eluate in 1 ml sequential aliquots in 10 microtubes.
- Using a Bradford assay, determine the protein concentration in each fraction.
- Apply the eluate that contains the fusion protein to a protein concentrator (100K MWCO).
- Centrifuge at 4 °C, 3,000 x g until the sample volume is reduced by 90%.
- Apply 4 ml protein storage buffer (see Recipes) and centrifuge at 4 °C, 3,000 x g, until the volume is reduced by 90%.
- Elute the protein on the filter with 300 μl protein storage buffer.
- Measure the protein concentration through Bradford assay with standard BSA in the spectrophotometer (the final concentration of SpCas9 should be approximately ~10 mg/ml).
- Transform 50 μl competent BL21-Pro cells with a pET28 vector containing 6xHis-SpCas9.
- Identify which samples from each step contain the fusion protein by SDS-PAGE.
- You should verify the activity of Cas9 protein using an in vitro cleavage assay.
- In vitro transcription of sgRNA
- Template extension
After template extension, purify DNA using a PCR purification kit.
Note: Purified DNA template can be stored at -20 °C.
Table 3. Oligos for in vitro transcriptionOligo F
(target specific)5’-GAAATTAATACGACTCACTATAG (target 20 nt - 5’ to 3’ direction, without PAM) GTTTTAGAGCTAGAAATAGCAAG-3’ Oligo R
(common )5’-AAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGAC
TAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3’ - In vitro transcription reaction

4 h-overnight reaction, 37 °C water bath or incubator. - Removal of DNA template
- Add 11 μl DNase I buffer
- Add 1 μl (2 U/μl) DNase I in 100 μl reaction
- Incubate at 37 °C, 30 min
- RNA purification
- Use an RNA cleanup kit following the manufacturer’s manual.
- Elute RNA in 10-25 μl DEPC treated water or RNase-free water. The final RNA concentration should be 5-15 μg/μl.
- Use an RNA cleanup kit following the manufacturer’s manual.
- Template extension
- Transfection of ribonucleoproteins (RNPs) to Chlamydomonas
- Preparation of cells and RNP complex for transformation
- Cultivate the Chlamydomonas cells mixotrophically (in the light with acetate) in 50 ml liquid TAP medium at 25 °C under continuous light (50-70 μmol photons m-2 sec-1) in a 100 ml flask with shaking at 90 rpm until the cell density reaches an OD750 of 0.3-0.5 as measured by a spectrophotometer.
- Estimate the cell density with a hemocytometer and microscope.
- Harvest 5 x 105 cells by centrifugation in the 1.5 ml Eppendorf tubes (2,000 x g, 3 min, room temperature), discard the supernatant, and resuspend the cells in 250 μl of TAP sucrose solution (see Recipes) by gently pipetting (prepare one aliquot of cells per transformation reaction).
- To prepare the RNP complex, premix purified Cas9 protein (200 μg) with in vitro transcribed sgRNA (140 μg) in the 1.5 ml Eppendorf tubes and incubate for 10 min at room temperature (prepare one aliquot of RNP complexes per transformation reaction).
- Add the RNP complex to the resuspended cells and gently tap the tube.
- Transfer 250 μl (+ volume of the RNP complex) of the transformation mixture to an electroporation cuvette.
- Incubate at room temperature for 5 min.
- Cultivate the Chlamydomonas cells mixotrophically (in the light with acetate) in 50 ml liquid TAP medium at 25 °C under continuous light (50-70 μmol photons m-2 sec-1) in a 100 ml flask with shaking at 90 rpm until the cell density reaches an OD750 of 0.3-0.5 as measured by a spectrophotometer.
- Electroporation of RNP complex
- Set parameters in the electroporation device (Bio-Rad Laboratories) (voltage: 600 V, capacity: 50 μF, resistance: infinity).
- Gently tap the cuvette to mix the contents and place the cuvette in the cuvette chamber.
- Electroporate the cells and then resuspend them by gently pipetting in 750 μl of TAP sucrose solution (total volume, 1 ml).
- Transfer all of the transformation mixture into a 15 ml conical tube.
- To obtain single mutant colonies, immediately transfer 2-4 x 103 cells (4-6 μl) from the transformation mixture to a new 15 ml conical tube and then plate them on a 1.5% TAP agar plate (see Recipes) using top agar (see Recipes). After transformation, 400-600 colonies generally appear (Figure 5).
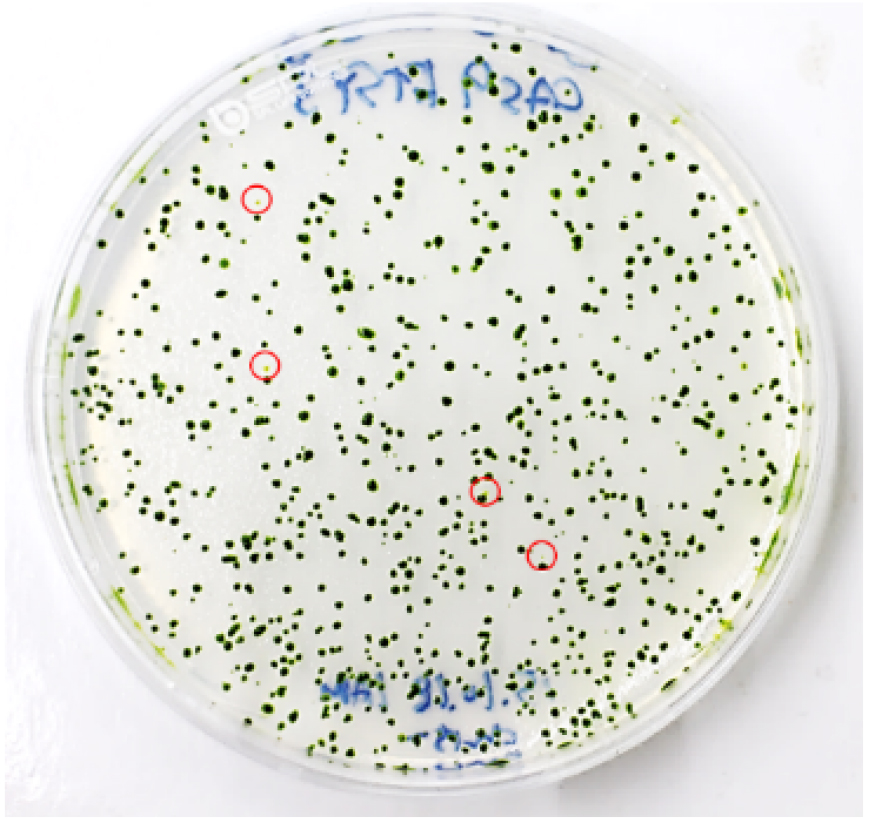
Figure 5. Visual coloration examination to investigate CpFTSY gene knockout. Red circles indicate the putative CpFTSY knockout mutant lines which have pale green colors grown on TAP agar medium. - Incubate the rest of the cells for 12 h in dim light (5-10 μmol photons m-2 sec-1), after which they should be harvested for genomic DNA extraction for use in targeted deep-sequencing analysis.
- Set parameters in the electroporation device (Bio-Rad Laboratories) (voltage: 600 V, capacity: 50 μF, resistance: infinity).
- Preparation of cells and RNP complex for transformation
- Preparation of NGS library using PCR
- Deep sequencing library preparation by PCR amplification
- (Optional) Target gene (~cleavage site ± 250 bp) amplification
For successful library construction, we recommend amplifying the target gene, which should include the cleavage site ± 250 bp, first. We use Phusion DNA polymerase (Thermo). - Pre-indexing PCR (~25 cycles)

For paired-end sequencing, the DNA library must have two adapters that include the i5 and i7 indices. Amplify the target DNA using Phusion polymerase with the pre-index tailed primers.
Note: We recommend that the length of the target DNA be ~250 bp for Miseq 300 cycles or ~450 bp for Miseq 500 cycles for optimal merging of the paired-end reads. - Indexing PCR (~25 cycles)

After the pre-indexed amplification, amplify the PCR products with universal index primers. Purify the amplicons using a PCR DNA purification kit and measure the concentration of library using a spectrophotometer.
- (Optional) Target gene (~cleavage site ± 250 bp) amplification
- Targeted deep sequencing
We perform targeted deep sequencing using an Illumina Miseq Reagent Kit (v2).
- Deep sequencing library preparation by PCR amplification
Data analysis
- Analysis of targeted deep-sequencing data
- We use Cas-Analyzer in RGEN tools (http://www.rgenome.net/cas-analyzer/) (Figure 6)
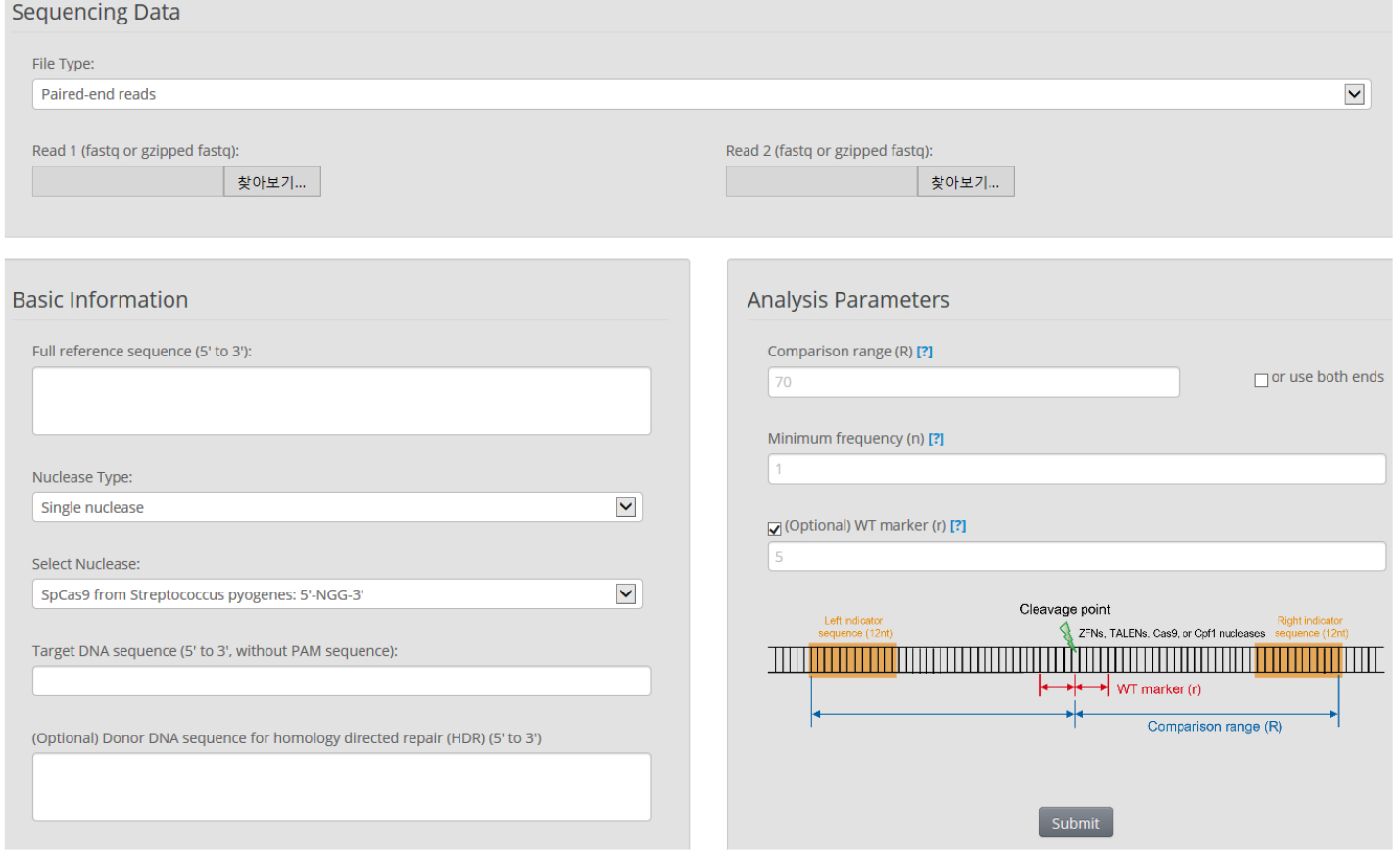
Figure 6. Overview of Cas-Analyzer. Users analyze their targeted deep-sequencing data using Cas-Analyzer without uploading any data to the server or local tool installation.
- Upload the NGS data files. Single-end reads, paired-end reads, and already merged sequencing data are allowed.
- Input the reference sequence and target DNA sequence without the PAM (protospacer adjacent motif) sequences that are the recognition site of CRISPR endonucleases.
- Submit the data.
- The results contain information about mutation counts and frequencies in brief. Statistical data and detailed sequence alignment data are displayed further down on the page.
- We use Cas-Analyzer in RGEN tools (http://www.rgenome.net/cas-analyzer/) (Figure 6)
- Analysis of off-target mutations in an isolated mutant
- Find more detailed information about potential off-targets associated with each selected target in the Chlamydomonas genome using Cas-OFFinder. Select a mismatch number (typically 4) and DNA/RNA bulge size (typically 1) (Figure 7).
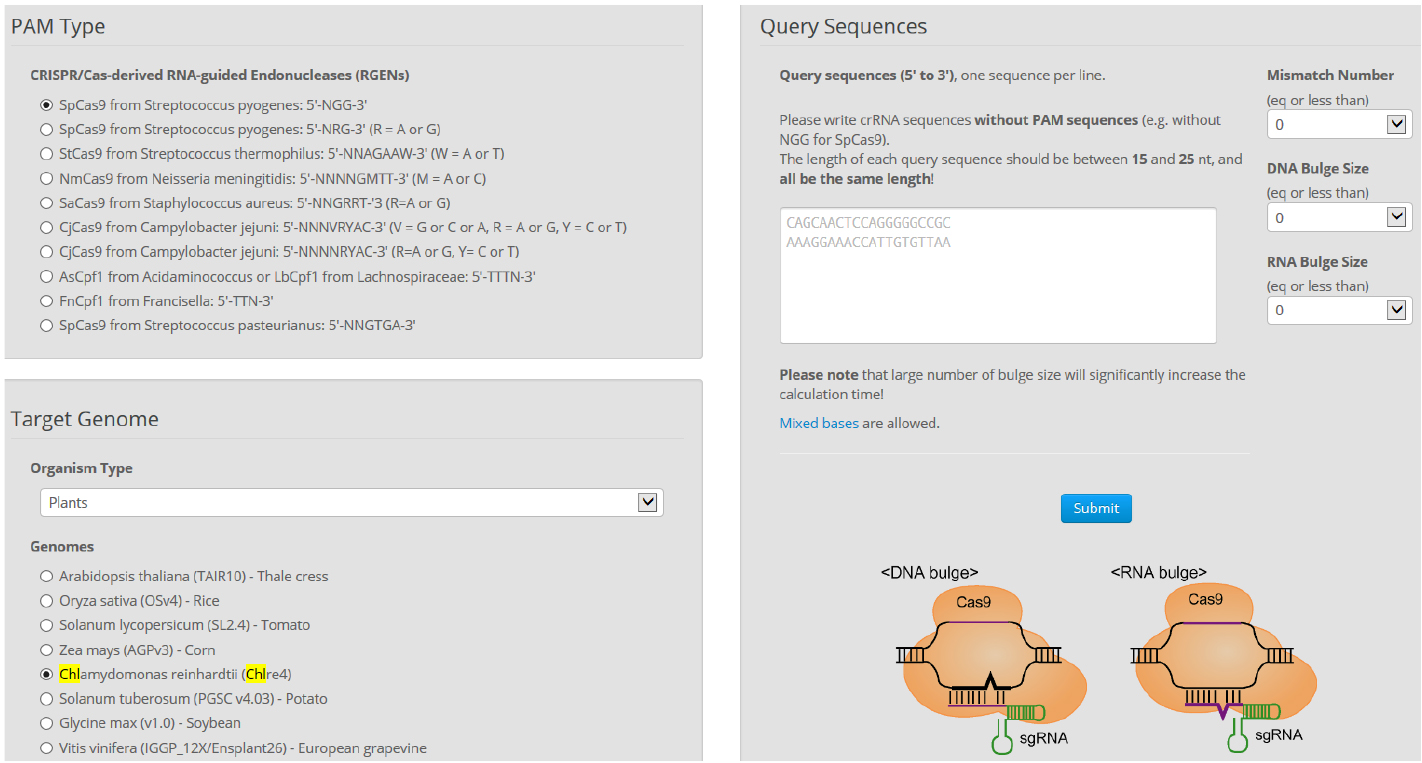
Figure 7. Overview of Cas-OFFinder. Cas-OFFinder (http://www.rgenome.net/cas-offinder/) has a broad range of searching options on the number of mismatches (10 bp) and DNA or RNA bulges (2 bp). - Genomic DNA preparation from wild-type cells and isolated mutants.
- For predicted off-targets, perform NGS library preparation, targeted deep sequencing (Procedure E), and NGS data analysis.
- Find more detailed information about potential off-targets associated with each selected target in the Chlamydomonas genome using Cas-OFFinder. Select a mismatch number (typically 4) and DNA/RNA bulge size (typically 1) (Figure 7).
Recipes
- Cas9 protein purification
- LB medium (for 1 L)
10 g NaCl
10 g tryptone
5 g yeast extract
Distilled water - 1 M IPTG stock
2.383 g of IPTG
10 ml distilled water
Filtered by syringe filter - Lysis buffer, pH 7.4 (10 ml for 200 ml E.coli culture)
50 mM NaH2PO4
300 mM NaCl
10 mM imidazole
Distilled water
Note: Adjust pH to 7.4 using NaOH, filtered. - Wash buffer, pH 7.4 (20 ml for 200 ml E.coli culture)
50 mM NaH2PO4
300 mM NaCl
20 mM imidazole
Distilled water
Note: Adjust pH to 7.4 using NaOH, filtered. - Elution buffer, pH 7.4 (10 ml for 200 ml E.coli culture)
50 mM NaH2PO4
300 mM NaCl
250 mM imidazole
Distilled water
Note: Adjust pH to 7.4 using NaOH, filtered. - Cas9 storage buffer, pH 7.5 (10 ml for 200 ml E.coli culture)
150 mM NaCl
20 mM HEPES
0.1 mM EDTA
1 mM DTT
2% sucrose
20% glycerol
Distilled water
Note: Adjust pH to 7.5 using NaOH, filtered. - Transfection
- TAP sucrose solution
TAP medium with 40 mM sucrose - TAP agar plates
TAP medium with 1.5% agar - Top agar
TAP medium with 0.5% agar
- TAP sucrose solution
Acknowledgments
This protocol was originally published as part of Baek et al. (2016). The authors especially thank Dr. Duk Hyoung Kim for developing the method. This work was supported by grants from the Korea CCS R&D Center (KCRC) (NRF-2014M1A8A1049273) to E.J. and the Plant Molecular Breeding Center of Next Generation BioGreen 21 Program (PJ01119201) to S.B.
References
- Baek, K., Kim, D. H., Jeong, J., Sim, S. J., Melis, A., Kim, J. S., Jin, E. and Bae, S. (2016). DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9 ribonucleoproteins. Sci Rep 6: 30620.
- Bae, S., Kweon, J., Kim, H. S. and Kim, J. S. (2014a). Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 11(7): 705-706.
- Bae, S., Park, J. and Kim, J. S. (2014b). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30(10): 1473-1475.
- Harris, E. H. (2001). Chlamydomonas as a model organism. Annu Rev Plant Physiol Plant Mol Biol 52: 363-406.
- Kim, S., Kim, D., Cho, S. W., Kim, J. and Kim, J. S. (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24(6): 1012-1019.
- Park, J., Bae, S. and Kim, J. S. (2015). Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31(24): 4014-4016.
- Park, J., Lim, K., Kim, J. S. and Bae, S. (2017). Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics 33(2): 286-288.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yu, J., Baek, K., Jin, E. and Bae, S. (2017). DNA-free Genome Editing of Chlamydomonas reinhardtii Using CRISPR and Subsequent Mutant Analysis. Bio-protocol 7(11): e2352. DOI: 10.21769/BioProtoc.2352.
Category
Plant Science > Plant transformation > Electroporation
Cell Biology > Cell engineering > CRISPR-cas9
Plant Science > Plant molecular biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.