- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Optogenetic Stimulation and Recording of Primary Cultured Neurons with Spatiotemporal Control
Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2335 Views: 12683
Reviewed by: Pengpeng LiZinan ZhouAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
We studied a network of cortical neurons in culture and developed an innovative optical device to stimulate optogenetically a large neuronal population with both spatial and temporal precision. We first describe how to culture primary neurons expressing channelrhodopsin. We then detail the optogenetic setup based on the workings of a fast Digital Light Processing (DLP) projector. The setup is able to stimulate tens to hundreds neurons with independent trains of light pulses that evoked action potentials with high temporal resolution. During photostimulation, network activity was monitored using patch-clamp recordings of up to 4 neurons. The experiment is ideally suited to study recurrent network dynamics or biological processes such as plasticity or homeostasis in a network of neurons when a sub-population is activated by distinct stimuli whose characteristics (correlation, rate, and, size) were finely controlled.
Keywords: Primary culture of neuronsBackground
Optogenetics provide a mean to control neuronal activity with millisecond precision. However, neurons are often activated simultaneously either by flashes of light that activate the whole population synchronously or by a light whose intensity is temporally modulated over the whole field of view (Boyden et al., 2005). Yet, several methods exist to modulate the light spatially and have been used to uncage glutamate (Nawrot et al., 2009) or activate channelrhodopsin (ChR2) expressing neurons (Guo et al., 2009) (for review of available methods to stimulate neurons with both spatial and temporal resolution see Anselmi et al., 2015).
To gain spatial control of the stimulation, a first possibility is to use a laser and move its beam quickly over different positions. For example, uncaging glutamate at different dendritic locations has been achieved by deflecting a laser beam with acousto-optic deflectors (Shoham et al., 2005). This strategy is likely viable only if we modulate the light intensity sufficiently slowly over a limited area. Alternatively, a spatial pattern of light can be achieved using phase or intensity light modulators. Holographic technique based on phase modulation permits to obtain an image in three dimensions with a good spatial precision but patterns can be displayed at a rate of only 100 Hz (Papagiakoumou et al., 2010). If a two dimensional pattern is sufficient, intensity modulation can simply be obtained by placing a projector or an array of LEDs in the conjugated plane of the sample (Farah et al., 2007; Guo et al., 2009). This technique has the advantages of being easy to implement, can target many regions of interest simultaneously and has the fastest temporal resolution.
Here we took advantage of a fast video projector based on the workings of a Digital Micromirror Device (DMD). A LED light source is split by an array of micromirrors that can be controlled with sub millisecond precision in order to display any arbitrary pattern of light (Barral and Reyes, 2016). An image of the projector is focalized to the sample plane via a pair of lenses and the microscope objective. The DMD technology offers an unprecedented temporal precision that enables to display patterns at 1.44 kHz and even faster DMDs are now available. In our settings, the resulting pixel size (2.2 x 1.1 µm) was sufficiently small to stimulate single neurons.
To activate a single neuron, we selected a region of interest of ~30 x 30 µm, centered at the soma of the neuron of interest and sent a 5 msec pulse of light. By designing patterns that are projected onto the sample, we could target independently and simultaneously a large number of neurons (10 to 100 neurons). Stimulated neurons were both excitatory and inhibitory (expression of ChR2 under the Synapsin promoter) and were activated by Poisson spike trains. The rate and correlation of the spike stimuli were controlled by the experimenter (see Barral and Reyes, 2016). By recording from neurons that expressed ChR2, we verified that stimulated neurons responded faithfully to the light pulses. We then recorded concurrently the membrane potentials of up to 4 neurons in cell-attached and in whole-cell configurations to isolate the spiking activity and the postsynaptic inputs, respectively.
Materials and Reagents
- For the neuronal culture
- Round coverslips, 25 mm diameter, German glass (Electron Microscopy Sciences, catalog number: 72196-25 )
- Siliconized low-retention microcentrifuge tubes (1.5 ml) (Fisher Scientific, catalog number: 02-681-331 )
- Low-retention pipet tips 200 µl (Fisher Scientific, catalog number: 02-717-165 )
- Low-retention pipet tips 1,000 µl (Fisher Scientific, catalog number: 02-717-166 )
- Disposable Petri dishes (35 x 10 mm) (Corning, Falcon®, catalog number: 351008 )
- Stericup-GP sterile vacuum filtration system, 0.22 µm, polyethersulfone (EMD Millipore, catalog number: SCGPU05RE )
- Syringe filters; MCE membrane; pore size: 0.22 µm (EMD Millipore, catalog number: SLGS033SS )
- Sterile transfer pipets (Fisher Scientific, catalog number: 13-711-20 )
- Conical sterile polypropylene centrifuge tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 339650 )
- Postnatal (P0-P1) mice
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: 320331 )
- Nitric acid (Sigma-Aldrich, catalog number: 258121 )
- 70% ethanol
- Poly-L-lysine hydrobromide–mol wt 70,000-150,000 (Sigma-Aldrich, catalog number: P1274 )
- Sodium tetraborate decahydrate ≥ 99.5% (Sigma-Aldrich, catalog number: B9876-500G )
- Boric acid (cell culture tested) (Sigma-Aldrich, catalog number: B9645-500G )
- HBSS (10x), no calcium, no magnesium, no phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 14185052 )
- Penicillin-streptomycin (10,000 U/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- HEPES (Thermo Fisher Scientific, GibcoTM, catalog number: 15630080 )
- Agar (Sigma-Aldrich, catalog number: A1296-100G )
- Sodium bicarbonate (NaHCO3) (Fisher Scientific, catalog number: S233 )
- Sodium pyruvate solution (Sigma-Aldrich, catalog number: S8636-100ML )
- Sodium hydroxide (NaOH) (Fisher Scientific, catalog number: S318 )
- L-cysteine hydrochloride monohydrate (Sigma-Aldrich, catalog number: C7880-500MG )
- DNase I grade II, from bovine pancreas–100 mg (Roche Diagnostics, catalog number: 10104159001 )
- Papain from Carica papaya–10 ml 100 mg (Roche Diagnostics, catalog number: 10108014001 )
- Calcium chloride (CaCl2) (Fisher Scientific, catalog number: C79 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266 )
- Trypsin inhibitor from chicken egg white (Sigma-Aldrich, catalog number: T9253-500MG )
- Albumin from bovine serum (powder, suitable for cell culture, = 96%) (Sigma-Aldrich, catalog number: A9418-5G )
- Trypan blue solution (0.4%, liquid, sterile-filtered, suitable for cell culture) (Sigma-Aldrich, catalog number: T8154-20ML )
- Neurobasal medium (1x) (Thermo Fisher Scientific, GibcoTM, catalog number: 21103049 )
- B-27 serum-free supplement containing vitamin A (50x), liquid (Thermo Fisher Scientific, GibcoTM, catalog number: 17504044 )
- GlutaMAXTM supplement (Thermo Fisher Scientific, GibcoTM, catalog number: 35050061 )
- AAV virus for ChR2 expression (University of North Carolina Vector Core Services, AAV2-hSyn-hChR2(H134R)-mCherry)
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: BP358 )
- D-glucose (Fisher Scientific, catalog number: BP350 )
- Potassium chloride (KCl) (Fisher Scientific, catalog number: P333 )
- Sodium phosphate monobasic (NaH2PO4) (Fisher Scientific, catalog number: S369 )
- K-gluconate (Sigma-Aldrich, catalog number: P1847 )
- Phosphocreatine (Sigma-Aldrich, catalog number: P1937 )
- ATP-Mg (Sigma-Aldrich, catalog number: A9187 )
- GTP (Sigma-Aldrich, catalog number: G8877 )
- Poly-L-Lysine (PLL solution) (see Recipes)
- Dissection solution (see Recipes)
- Papain solution (see Recipes)
- DNase/L-cysteine solution (see Recipes)
- DNase/Mg solution (see Recipes)
- Trypsin inhibitor solution (see Recipes)
- Culture medium (NB/B27 medium) (see Recipes)
- Round coverslips, 25 mm diameter, German glass (Electron Microscopy Sciences, catalog number: 72196-25 )
- For electrophysiology recordings
- Borosilicate glass capillaries (1.5 OD) (World Precision Instruments, catalog number: 1B150F-4 )
- Artificial cerebrospinal fluid (aCSF, see Recipes)
- Intracellular solution (see Recipes)
- Borosilicate glass capillaries (1.5 OD) (World Precision Instruments, catalog number: 1B150F-4 )
Equipment
- For the neuronal culture
- Tissue culture hood
- P1000 pipet
- P200 pipet
- Cell culture incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: FormaTM Series II 3110 , catalog number: 3110)
- Vibratome slicer (Leica Biosystems, model: Leica VT1200 S )
- Dumont #5 forceps (Fine Science Tools, catalog number: 11251-30 )
- Dumont #7 forceps (Fine Science Tools, catalog number: 11271-30 )
- Extra fine bonn scissors (straight) (Fine Science Tools, catalog number: 14084-08 )
- Tissue culture hood
- For the electrophysiology setup
- Upright water immersion microscope with fluorescence (Olympus, model: BX51 )
- Fluorescence filter set for mCherry (TRITC, Chroma Technology, model: 41002c )
- CCD camera for fluorescence imaging (Hamamatsu Photonics, model: C8484 )
- CCD camera for IR imaging (Olympus, model: OLY-150 IR )
- Micromanipulator (Luigs & Neumann, model: SM6 )
- Patch-clamp amplifiers (Dagan, model: BVC-700A )
- 18-bit interface card (National Instruments, model: PCI-6289 )
- Flaming/Brown micropipette puller (Sutter Instrument, model: P-97 )
- Hemacytometer (Hausser, catalog number: 3110 )
- Upright water immersion microscope with fluorescence (Olympus, model: BX51 )
- For the optogenetic setup
- DLP projector (Texas Instruments, model: DLP® LightCrafterTM Evaluation Module )
- Aluminum Breadboard, 150 x 300 x 12.7 mm, double density, M6 thread (Thorlabs, model: MB1530/M )
- Ø1" Achromatic Doublet, SM1-Threaded Mount, f = 35 mm, ARC: 400-700 nm (Thorlabs, model: AC254-035-A-ML )
- Ø2" Achromatic Doublet, SM2-Threaded Mount, f = 200 mm, ARC: 400-700 nm (Thorlabs, model: AC508-200-A-ML )
- Ø2" (Ø50.8 mm) Protected Silver Mirror, 0.47" (12.0 mm) Thick (Thorlabs, model: PF20-03-P01 )
- Kinematic Mount for Ø2" Optics (Thorlabs, model: KM200 )
- U-DP; Dual port intermediate tube (Olympus, model: U-IT140 )
- U-EPA2; Eyepoint adjuster, BX2, Raises eyepoint 30MM (Olympus, model: U-IT101 )
- BX2 filter cube (U-MF2) for Olympus BX2 and IX2 models (Chroma Technology, model: U-MF2, catalog number: 91018 )
- 510 nm beamsplitter (BS, Part Size: 25.5 x 36 x 1 mm) (Chroma Technology, model: T510lpxrxt )
- 50/50 beamsplitter (BS, Part Size: 25.5 x 36 x 1 mm) (Chroma Technology, model: 21000 )
- EVGA GeForce GT 520 graphic card with a mini HDMI connector (EVGA, model: 01G-P3-1526-KR )
- Light-to-voltage optical sensors (ams, TAOS, model: TSL13T )
- DLP projector (Texas Instruments, model: DLP® LightCrafterTM Evaluation Module )
Procedure
- Primary cultures of neurons expressing channelrhodopsin
Note: Protocol for the culture of primary neurons was originally described by (Brewer et al., 1993) and was further modified by Hilgenberg and Smith (2007) and Barral and Reyes (2016). This procedure requires aseptic conditions for which the Gibco Cell Culture Basics Handbook can provide a good introduction.- Coating of the coverslips (prepare the day before culturing under sterile conditions)
- Clean the coverslips in 3 N HCl or concentrated nitric acid (70% wt/wt) for at least 24 h.
- Rinse the coverslips 3 times in distilled H2O, clean them with 70% ethanol and let dry under the tissue culture hood.
- Coat cleaned coverslips with 0.1 ml of PLL solution (see Recipes) for 12 h.
- Rinse 3 times in distilled H2O and let dry.
- Dissociation and culture of primary neurons. Depending on the size of the slicing container, 100 to 150 ml of dissection solution is needed (see Recipes).
- Cut 5-6 600 µm coronal slices of cortex from postnatal (P0-P1) mice in ice-cold dissection solution.
- In the meantime, prepare the papain solution (see Recipes).
- Using forceps, remove the thin membrane covering the brain called the meninges. Dissect out the cortex and cut the cortical layer in small pieces of 0.5-1 mm in length. Transfer the brain pieces in clean dissection solution.
- From this step on, the procedure has to be performed under a tissue culture hood. Filter the papain solution in a Petri dish and incubate the brain pieces in the papain solution for 20 min at 37 °C.
- In the meantime, dilute the DNase/Mg solution (see Recipes) in 10 ml and prepare 4 conical tubes as follow:
1: 4 ml of DNase/Mg solution
2: 2 ml of DNase/Mg solution + 200 µl of trypsin inhibitor solution (see Recipes)
3: 2 ml of DNase/Mg solution + 60 µl of trypsin inhibitor solution
4: 2 ml of DNase/Mg solution + 30 µl of trypsin inhibitor solution - Using the transfer pipet, transfer the tissue in tube 1 and let the pieces settle down. Try to transfer the brain tissue with the smallest possible volume of solution. Repeat with tubes 2-4.
- Transfer the pieces in about 0.6 ml of equilibrated culture medium (see Recipes) in a low retention tube. Pipet 4-5 times with a P1000 low retention pipet and let the large pieces settle down. Transfer the pellet to a second low retention tube containing about 0.6 ml of culture medium. Pipet 4-5 times with a P200 low retention pipet and pool the two tubes.
- Dilute 10 μl of cell suspension in 10 μl of 0.4% trypan blue and estimate the number of viable cells using a hemacytometer. This is useful to control the final density of neurons.
- Plate 0.2-3 x 106 cells on each coverslip, resulting in a density of ~50-1,000 cells/mm2 at the time of experiment. Let settle for 1 h (in the incubator) for cells to adhere before flooding the Petri dish with 3 ml of equilibrated culture medium. Maintain cultures at 37 °C and 5% CO2.
- Exchange one third of the medium every 2-3 days. Cultures can be maintained in these conditions for up to 3 weeks (see Figure 1 for representative results).
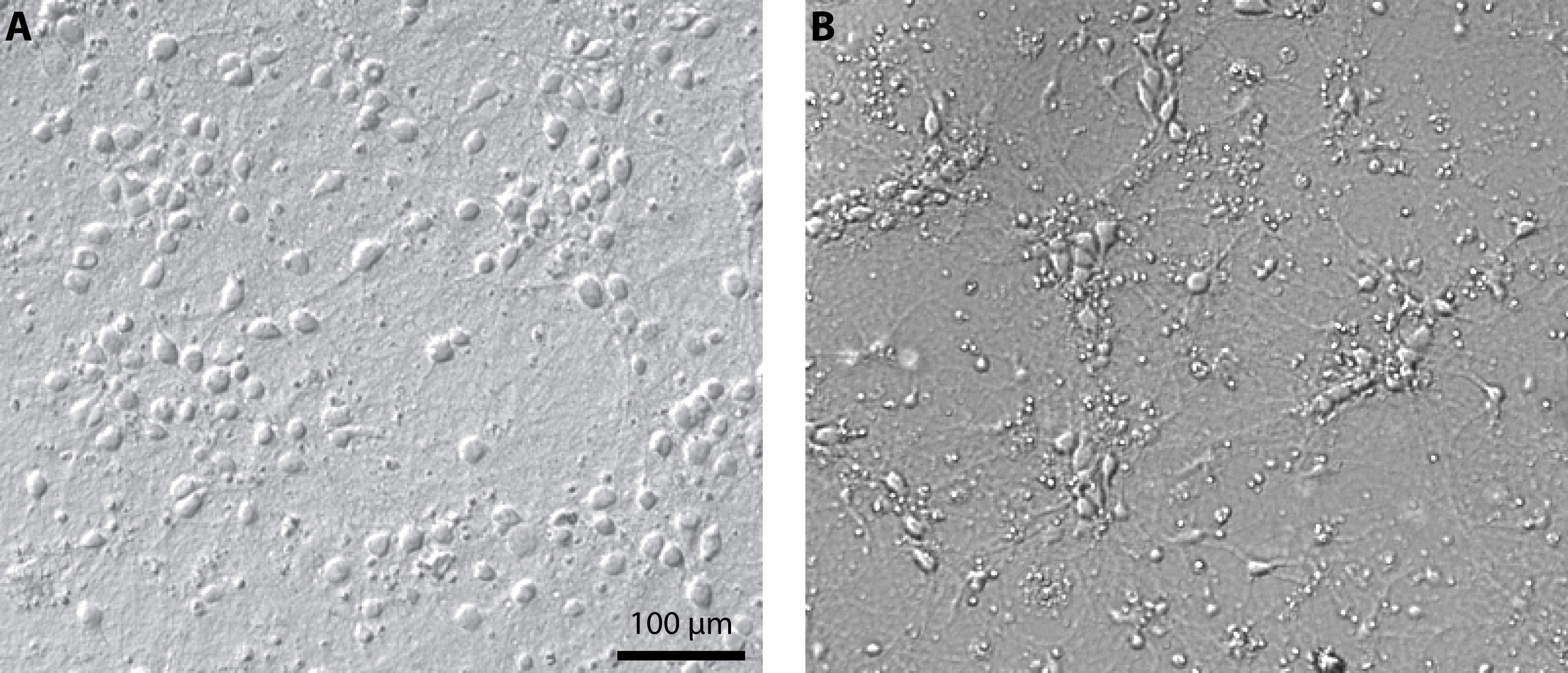
Figure 1. Culture of primary cortical neurons at 14 days in vitro. Representative examples of a healthy culture where cell bodies are well defined (A) and a culture where the coverslip coating was not achieved properly which resulted in cell aggregates and debris (B). - Expression of channelrhodopsin
- The virus AAV2-hSyn-hChR2(H134R)-mCherry) was produced at 3 x 1012 cfu/ml by the University of North Carolina Vector Core Services using plasmid generously provided by Karl Deisseroth (Stanford University). The virus could also be produced in house using standard protocol (Luo et al., 2007).
- Infect each Petri dish with 1 µl of virus after 3 days in vitro.
- Experiments can be performed between 14-21 DIV, when neuronal characteristics and network connectivity were stable and expression of ChR2 was sufficient to enable reliable photostimulation.
- The virus AAV2-hSyn-hChR2(H134R)-mCherry) was produced at 3 x 1012 cfu/ml by the University of North Carolina Vector Core Services using plasmid generously provided by Karl Deisseroth (Stanford University). The virus could also be produced in house using standard protocol (Luo et al., 2007).
- Coating of the coverslips (prepare the day before culturing under sterile conditions)
- Building and calibrating the photostimulation setup
The setup for optical stimulation using a video projector was originally described in (Stirman et al., 2012) and further modified in (Barral and Reyes, 2016). The reader will find much details of the procedure to combine a video projector with the microscope in (Stirman et al., 2011; Stirman et al., 2012). The general idea is to focus the image of the projector in the same plane as the sample. Because our microscope was mounted on a movable platform, the photostimulation setup had to be physically connected to the microscope. We describe here the procedure to achieve this but alternative solutions are available and might be more appropriate depending on the setup configuration. Here, we attached a breadboard onto the fluorescent path of the microscope to build the photostimulation light path. We chose the breadboard small enough to limit weight on the microscope but it forced us to use an additional mirror. If possible, try to avoid this mirror by having a longer straight path to project the light coming from the projector.- Building the stimulation setup
- Attach the dichroic mirror DM1 to the cube and insert it in the dual port. Place the eyepoint adjuster and the dual port above the fluorescent path and below the camera tube lens of the microscope (Figure 2A).
- Fix the aluminum breadboard on the fluorescent tube and define the optical path. Make use of a reflective mirror (Figure 2A, RM) if necessary.
- Place the Ø2" lens that will play the role of the tube lens (Figure 2B, PTL) as close as possible to the dichroic mirror and evaluate the position of the primary image plane.
- Place the DLP projector and the Ø1" zoom lens (Figure 2B, ZL) to form an image of the projector exactly at this location. It might be useful to mount the projector tube lens on a movable track in order to finely adjust its position when the microscope objective is changed for example.
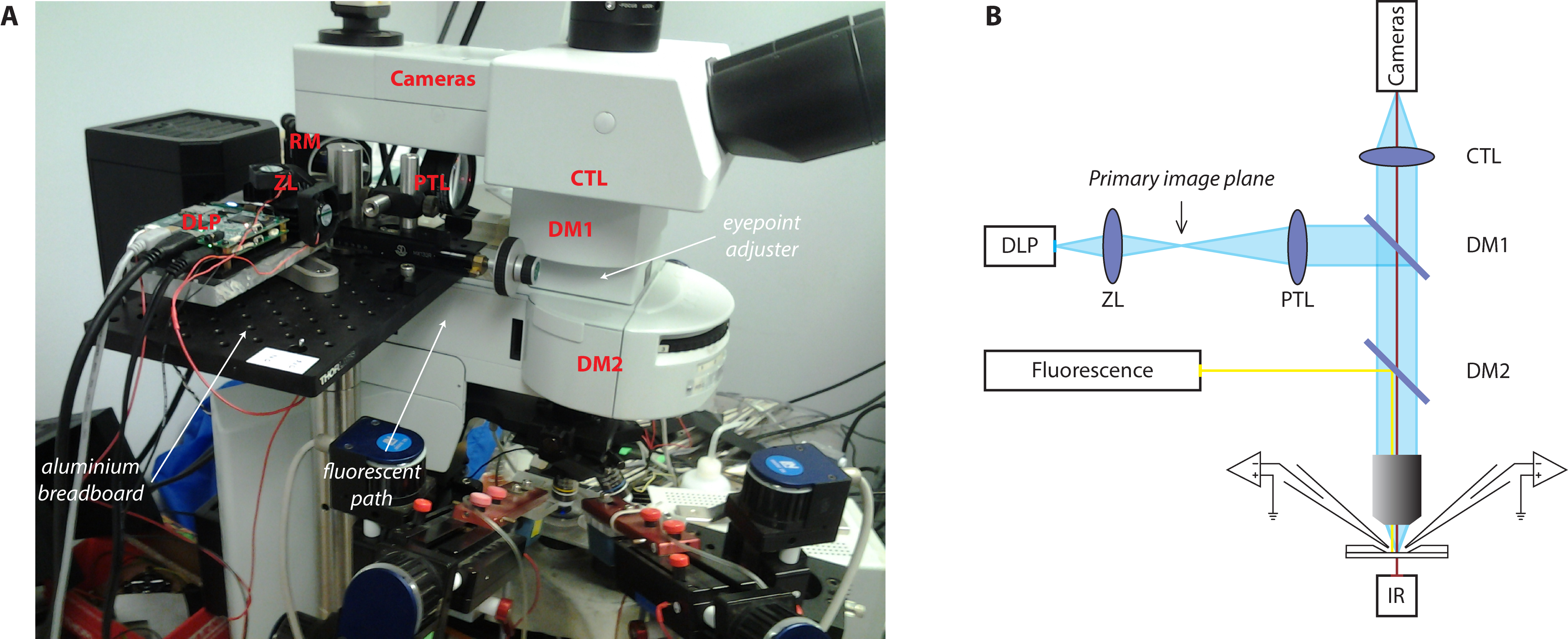
Figure 2. Photostimulation setup. A. Picture of the photostimulation setup; B. Schematic of the light path. DLP, Digital Light Processing projector; ZL, zoom lens; PTL, projector tube lens; EPA, eyepoint adjuster; CTL, camera tube lens; DM, dichroic mirror; RM, reflective mirror. - It is likely that the collimated beam will be larger than the dual port entry of the microscope. Install the photodiode next to the entry port of the microscope (Figure 3) to collect some of this light from the projector that will be used as a trigger to synchronize photostimulation and electrophysiological recordings. If the light beam is smaller than the entry port, place a 10/90 beamsplitter in-between the projector tube lens (PTL) and the dichroic mirror (DM1) to collect some light from the projector and redirect it to the photodiode system.

Figure 3. Photodiodes system. The photodiodes are placed next to the entry port of the microscope and collect a fraction of the beam coming from the projector (dashed line). - For recording of neuronal activity, we used patch-clamp recordings of up to 4 neurons in cell-attached and whole-cell configurations to monitor spikes and post-synaptic potentials, respectively. A useful summary of patch-clamp procedure can be found on Axolbio webpage. Alternatively, the setup is fully compatible with the use of microelectrode array (MEA) recordings if many neurons need to be recorded. Different commercial MEA systems are available or an open-source DIY system called NeuroRighter can be built for a lower cost.
- Attach the dichroic mirror DM1 to the cube and insert it in the dual port. Place the eyepoint adjuster and the dual port above the fluorescent path and below the camera tube lens of the microscope (Figure 2A).
- Calibrating the setup
- Brightness and pixel size
- Project a full-on pattern and measure the total light flux using a light power meter (Figure 4A).
- Measure the area of projection using a calibrated camera to deduce the resulting pixel size and the light intensity in mW/mm2. With the maximum current to the blue LED, we achieved 15 mW/mm2.

Figure 4. Calibration of the photostimulation setup. Images sent to the projector (left) and resulting images from the camera (right) when a full-on (A), ANSI checkerboard (B), or a 4 x 4 white dots (C) pattern were projected.
- Project a full-on pattern and measure the total light flux using a light power meter (Figure 4A).
- Contrast ratio
Here we want to measure the light intensity at the point of stimulation and at other locations to estimate the contrast ratio. This measure is important because it provides an estimate of the light intensity that a non-stimulated neuron receives during stimulation of ChR2 expressing neurons.- Place a 50/50 beamsplitter at the place of DM1 and place a 100% reflecting mirror at the sample plane and use the camera to measure the pixel intensity.
- Use a camera to measure the average pixel brightness of a white and of a black test pattern. Estimate the contrast ratio using the full-on full-off method measurements as a ratio of white to black. We measured 815:1. Use the faintest LED current for this procedure to avoid saturation of the camera.
- Project an ANSI checkerboard pattern composed of 16 rectangles, eight white and eight black (Figure 4B). Measure the ANSI contrast ratio as the quotient of the averaged white pixels to the black pixels. We measured 21:1, compared to the 43:1 value that was provided by the manufacturer of the projector. It gives a lower bound of the contrast ratio since a large amount of pixels (half of them) are ON in this configuration.
- To measure the real contrast ratio during experiment, namely to estimate background illumination of the system, we measured the contrast ratio when a single region of interest was illuminated or when 20 areas were simultaneously illuminated. We found the respective values of 700:1 and 170:1. During an actual experiment with a light intensity of 10 mW/mm2, the background light intensity is therefore between 10 and 50 µW/mm2, which would give rise to photocurrent of about 8-32 pA. This current is not sufficient to evoke spikes but can still elicit postsynaptic-like potentials on the order of 0.1 to 1 mV. However, the largest estimate of background stimulation is only relevant when neurons are activated simultaneously.
- Place a 50/50 beamsplitter at the place of DM1 and place a 100% reflecting mirror at the sample plane and use the camera to measure the pixel intensity.
- Relation between projector and camera pixels
- Project a pattern of 4 x 4 white dots evenly spaced on a black background (Figure 4C).
- Measure their positions on the camera.
- Knowing the position of the dots on the projected pattern and their locations on the image from the camera, use interpolations techniques to map every pixel of the camera to every pixel of the projector.
- This procedure can be achieved either by placing a mirror at the sample plane. One can also simply look at the reflection of the projector on the glass coverslip of the sample, which is useful if one wants to run this calibration during an actual experiment.
- Draw an arbitrary ROI, build the corresponding pattern, project and image it to assess the quality of the calibration.
- Project a pattern of 4 x 4 white dots evenly spaced on a black background (Figure 4C).
- Brightness and pixel size
- Interfacing the projector and the computer
The projector was controlled by the computer through a USB-based connection. One display mode (‘pattern sequence’ mode) enabled displaying a limited number of single-bit pattern at a rate up to 4 kHz. With the DLP lightcrafter, only 96 patterns can be stored. In this configuration, the predefined patterns were loaded into the remote memory of the projector and displayed using an external analog trigger. More recent projectors can have additional memory but it will never be sufficient to display patterns at high rate for more than few seconds. Additionally, the time to transfer data to the projector can be prohibitive.
To increase the number of patterns, an alternative method (‘HDMI video’ mode) was used to stream the images through a high speed HDMI connection. Here, images were sent continuously from the computer to the projector via the graphic card using the HDMI port. We used the ‘monochrome 1-bit per pixel’ video mode of the projector. This mode allows bypassing any video processing algorithms. A 24-bits RGB image is then considered as a succession of 24 single-bit planes. The resulting time resolution becomes 24 x 60 Hz = 1,440 Hz for a single-bit image, but without any limitation on the number of displayed patterns.
Synchronizing accurately the display of each frame with the acquisition of electrophysiological signals was a complicated task because it depends on the precise time at which the computer sends an image to the projector. Since the trigger present on the video projector was already used as an input trigger for the ‘pattern sequence’ mode, we chose to directly measure the light intensity at the output of the projector using a photodiode. Because, the collimated beam was slightly larger than the entry port of the microscope, we could place the photodiode at the edge of the aperture to capture light that did not enter the microscope (see Figure 3). Due to hardware specifications, there is a small 135 µsec delay for every 24-bits plane (i.e., every 16.7 msec) where the image becomes completely dark. We took advantage of this delay, which serves as an accurate trigger for synchronizing photostimulation and electrophysiological recordings offline.- Install the graphic card.
- Install the Graphical User Interface (GUI) provided by Texas Instrument and follow the instructions. Connect the projector using the USB and the HDMI connectors.
- Define the projector as an external display of the computer.
- Install the graphic card.
- Building the stimulation setup
- Independent stimulation of neurons using patterned illumination and recording of neuronal activity
- Transfer the neuronal culture coverslip on the microscope chamber and flood with artificial cerebrospinal fluid (see Recipes).
- Take a fluorescent image of the neuronal culture (Figure 5).

Figure 5. Pattern stimulation of the neuronal culture. 36 regions of interest (ROIs) are drawn around stimulated neurons (blue squares). 3 patch-clamp electrodes record spikes and/or subthreshold membrane potential of non-stimulated neurons. - Define regions of interest (ROIs) onto neurons that will be activated (Figure 5).
- Using a custom-built interface (Labview or Matlab are suitable for this task), build a temporal sequence of images in order to stimulate each ROI with the appropriate temporal pattern (see Table 1 for the key elements of the interface).
Table 1. Key elements that the custom-built interface must be able to achieve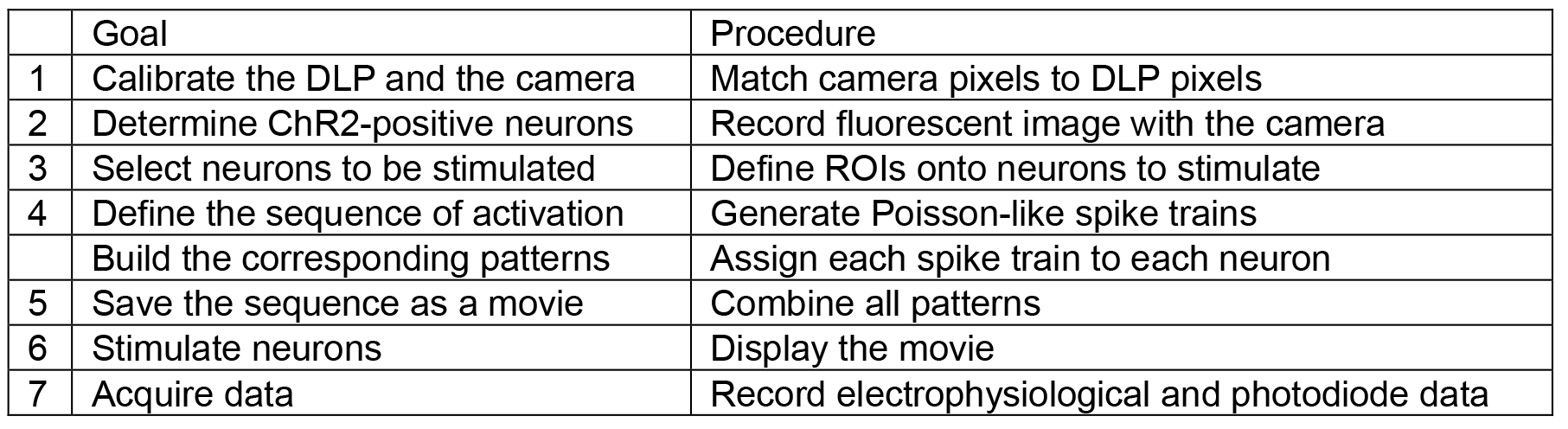
- Compile a movie from this sequence of images.
Note: Remember to save the movie without any compression since it would destroy the desired pattern sequence. - Pull recording pipets (aim for a pipet resistance of 5-8 MΩ) and fill patch-clamp electrodes with the internal solution (see Recipes).
Note: Here recordings were made in current-clamp configuration, therefore a high pipet resistance was acceptable and had the advantage of being easier to form a seal. - Select a neuron. Patch the neuron and record the electrophysiological signal in cell-attached or whole-cell configuration.
Note: Add QX-314 (Na+ channel blocker) to block spikes if excitatory or inhibitory postsynaptic potentials need to be isolated. - Display the stimulation movie through the projector (see Video 1) and record both the electrophysiological signal and the light intensity from the photodiode. Here, signals were filtered at 5 kHz and digitized at 25 kHz using an 18-bit interface card.
 Video 1. Pattern stimulation of the neuronal culture. 36 regions of interest (ROIs) are drawn around stimulated neurons (blue squares). Each stimulated neuron is activated by a train of light pulses (the actual stimulus is shown in blue on the top left corner). Three neurons are recorded simultaneously in cell-attached mode (yellow raster plots of evoked spikes in each corner, 15 repetitions of the stimulus) and then in whole-cell configuration (yellow trace of membrane potential, average of 7 repetitions).
Video 1. Pattern stimulation of the neuronal culture. 36 regions of interest (ROIs) are drawn around stimulated neurons (blue squares). Each stimulated neuron is activated by a train of light pulses (the actual stimulus is shown in blue on the top left corner). Three neurons are recorded simultaneously in cell-attached mode (yellow raster plots of evoked spikes in each corner, 15 repetitions of the stimulus) and then in whole-cell configuration (yellow trace of membrane potential, average of 7 repetitions). - Using the data from the photodiode, determine the precise time point where the light stimulation actually started. Realign the electrophysiological to this true starting point and analyze data offline.
- Transfer the neuronal culture coverslip on the microscope chamber and flood with artificial cerebrospinal fluid (see Recipes).
Data analysis
- Control experiment
To assess the effectiveness of the photostimulation setup in evoking spikes, we stimulated ChR2-positive neurons. We verified that spikes were evoked reliably when a single neuron was stimulated (Figure 6A). Then we stimulated 11 neurons simultaneously and recorded spikes from 2 of them (Figure 6B). Whereas one neuron responded only when stimulated by the light pulses, the other neuron also displayed action potentials in response to the stimulation of neighboring neurons, meaning that it was an integrant component of the recurrent network.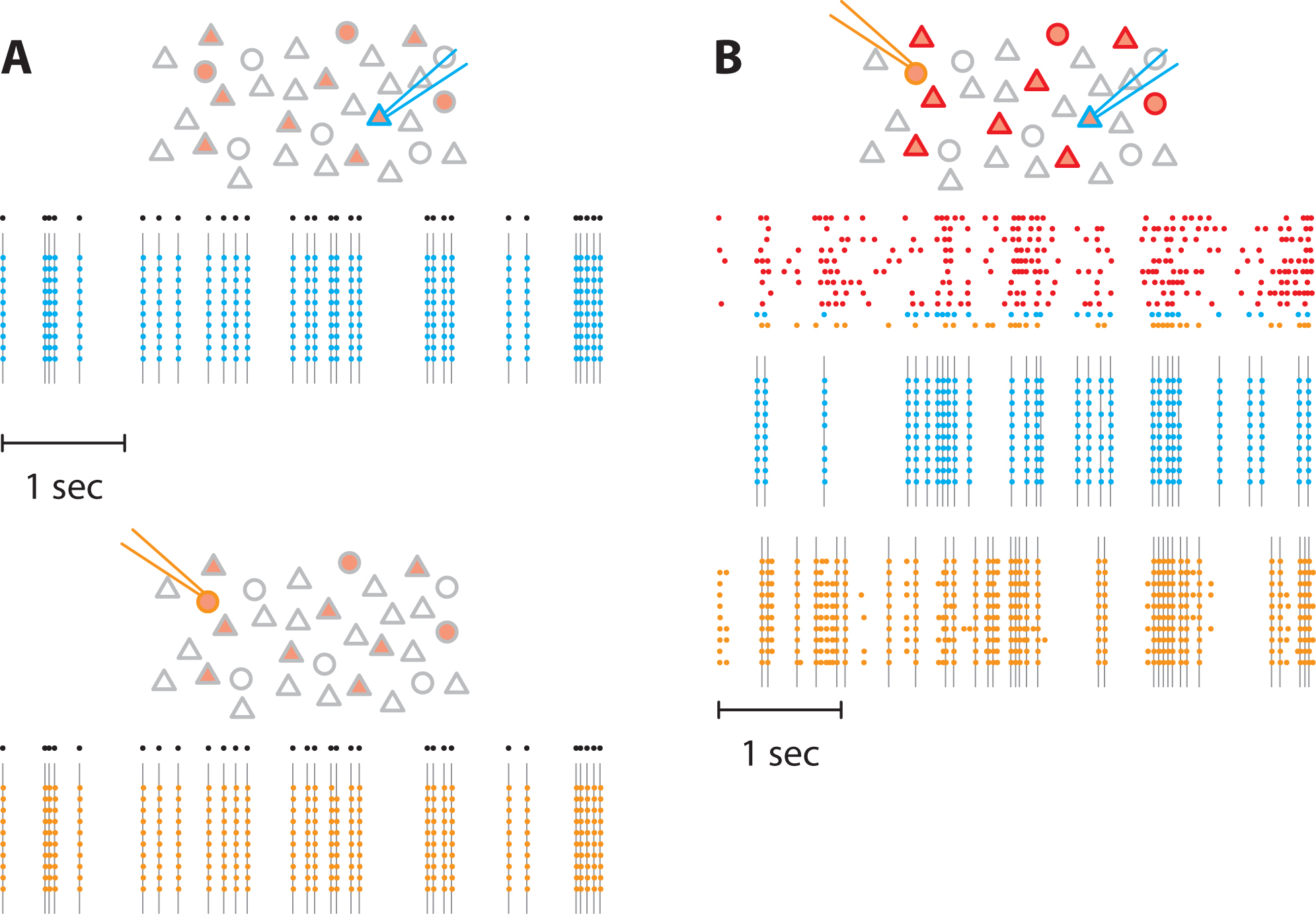
Figure 6. Pattern stimulation efficacy in evoking spikes in ChR2-expressing neurons. In the following schematics, neurons expressing ChR2 are shown in light red. Recorded neurons are designated by the recording pipets. Light-stimulated neurons have colored contours. A. A train of action potentials (black dots) was applied to two different neurons expressing ChR2 (blue and orange) independently (top and bottom, respectively). Several trials were realized to confirm the faithful stimulation of neurons of interest. Colored dots denote recorded action potential and grey lines the light flashes. No action potentials were observed in the non-stimulated neuron. B. Then, we stimulated 11 neurons in the network. We generated 11 spike trains (top) that were applied to neurons expressing ChR2, including the two previous neurons. Here, we used correlated spike trains for the stimulus (the average correlation between spike trains was C = 0.5). The blue and the orange neurons were recorded simultaneously. - Network stimulation
The final experiment consists in stimulating the selected neurons by a controlled stimulus (Figure 7A). Here, the firing rate of the input was fixed at 5 Hz and the spike trains were not correlated. These quantities can be varied at will by the experimenter. Neuronal activity in non-stimulated neurons was monitored either in cell-attached (Figure 7B) or in whole-cell (Figure 7C) configuration to isolate the spiking activity and the postsynaptic inputs, respectively. Cell-attached recordings (Figure 7B) showed that some cells fired robustly (neuron 2) while little activity was evoked in others (neurons 1 and 3). Subsequent whole-cell recordings in the same neurons also showed that large voltage transients evoked reliable action potentials across trials. The amplitudes were much larger than the unitary EPSPs and were mostly likely due to synchronous recurrent activity (Barral and Reyes, 2016).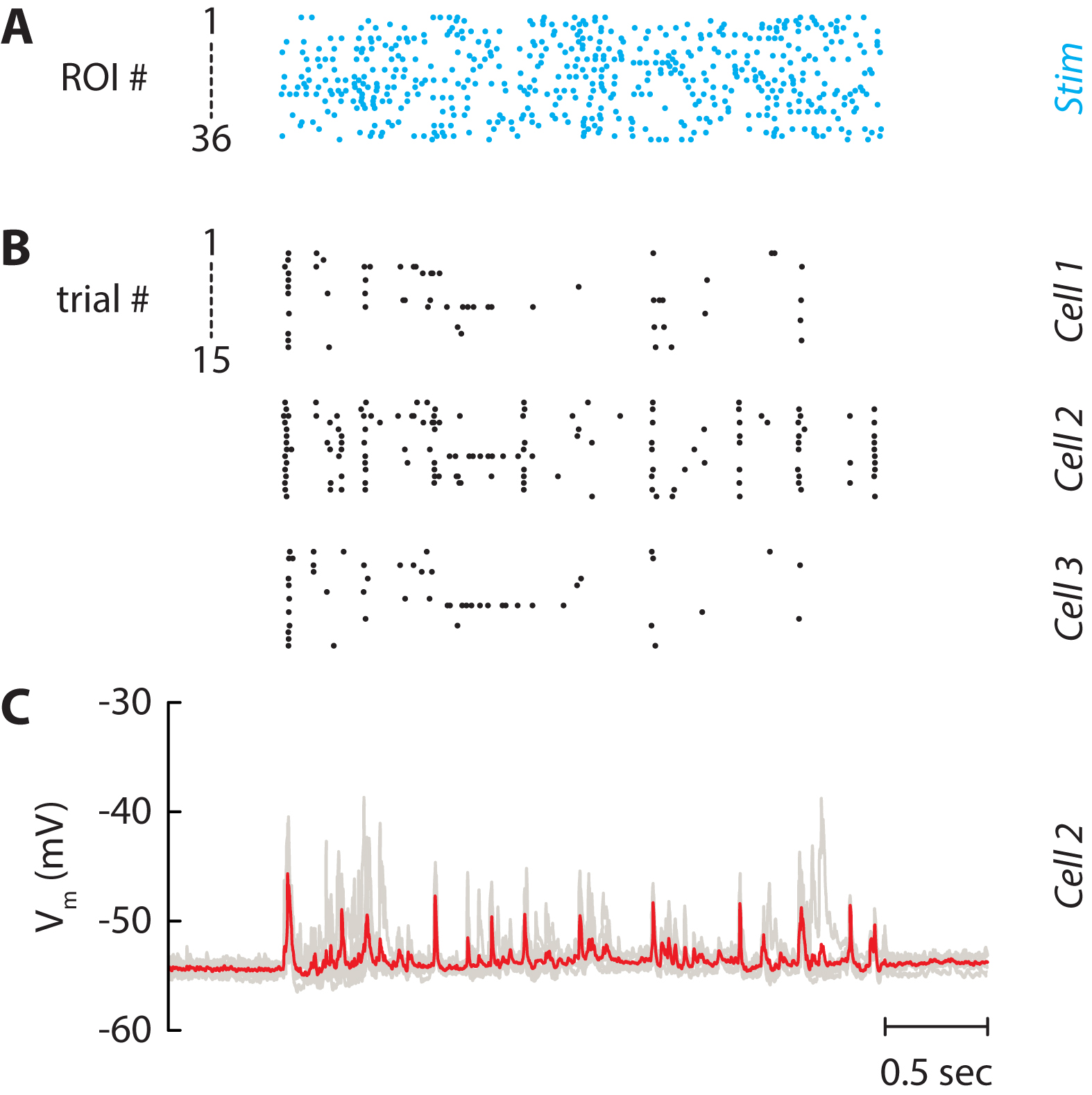
Figure 7. Independent stimulation of the neuronal network. A. Spike trains which are used for stimulating the 36 selected neurons in Figure 5. Each line is a train of light pulses applied to a given ROI. B. Raster plot of three neurons recorded simultaneously. Each line represents a given repetition of the same stimulus (15 repetitions). C. Corresponding membrane potential of Cell 2. Each repetition (7 repetitions) is drawn as a grey line and the average is displayed in red. The patch pipet contained the Na+ channel blocker QX-314 to block spikes and isolate postsynaptic potentials.
Notes
- The protocol to culture primary cortical neurons can work equally on hippocampal neurons.
- Add 5 μM Ara-C or 5 μM 5-Fluoro-2’-deoxyuridine on day 3-5 if glial cells show excessive proliferation. However, it is usually not necessary since Neurobasal culture medium with B-27 supplement does not promote glia cell proliferation and it might increase cell death.
- We described here a setup using a 460 nm light source to stimulate ChR2-expressing neurons. The setup can easily be modified to use another LED of the projector. Alternatively, the light engine of the projector can be dismounted and an external light source can be used to enlighten the digital micromirror device.
Recipes
- Poly-L-Lysine (1x, 0.1 mg/ml PLL, 20 aliquots of 0.5 ml)
- Make 100 ml of boric acid/sodium tetraborate solution (0.1 M, pH 8.4)
Dissolve 0.95 g sodium tetraborate decahydrate in 100 ml ddH2O
Add boric acid to borax solution until desired pH (pH 8.4) is reached (about 0.61 g of boric acid) - Dissolve 1 mg of PLL in 10 ml boric acid/sodium tetraborate solution
- Filter through 0.2 μm, make 0.5 ml aliquots and store at -20 °C for up to 6 months
- Make 100 ml of boric acid/sodium tetraborate solution (0.1 M, pH 8.4)
- Dissection solution (1x, CMF-HBSS/HEPES with antibiotics, 1 L)
Note: Ca2+ and Mg2+ free Hank’s balanced salt solution containing 1 mM pyruvate,15 mM HEPES, 10 mM NaHCO3 and antibiotics. - Mix
100 ml of 10x CMF-HBSS
3.9 g HEPES (15 mM final)
0.84 g NaHCO3 (10 mM final)
10 ml 100x sodium pyruvate (1 mM final)
10 ml 100x penicillin/streptomycin (100 U/ml penicillin/100 μg/ml streptomycin final)
800 ml of distilled H2O - Adjust pH to 7.2 with 1 N NaOH and add H2O to 1 L
- Filter sterilize through 0.2 μm sterile filter, store up to 1 month at 4 °C if the pH remains stable
- Papain solution (1x, 15 U/ml papain, 100 U/ml DNase, 2 ml)
- Add 30 U papain (i.e., 100 µl) to 1.8 ml of dissection solution and warm up to 30-32 °C for 20 min to clear the solution before use
- Add 0.2 ml of DNase/L-cystein solution (containing 200 U of DNase and 0.4 mg of L-cysteine)
- Adjust pH to 7.4 with about 5 μl of 0.1 N NaOH
- Filter through 0.2 μm and use it right away
- DNase/L-cysteine solution for papain solution (20x, 100 U/ml DNase final, 1.8 mM L-cystein final, 10 aliquots of 0.2 ml)
- Dissolve 2,000 U (1 mg) of DNase and 6.4 mg of L-cysteine in 2 ml of dissection solution
- Make 0.2 ml aliquots and store at -20 °C for up to 6 months
- DNase/Mg solution for dissociation (50x, 20 U/ml DNase final, 10 aliquots of 0.2 ml)
- Dissolve 2,000 U (1 mg) of DNase, 81.3 mg of MgCl2 (2 mM final) and 14.7 mg CaCl2 (1 mM final) in 2 ml of dissection solution
- Filter through 0.2 µm, make 0.2 ml aliquots and store at -20 °C for up to 6 months
- Trypsin inhibitor solution (100 mg/ml BSA, 40 mg/ml trypsin inhibitor)
- In 3 ml of dissection solution (or CMF-HBSS), dissolve 0.3 g BSA (Bovine Albumin) and 0.12 g of trypsin inhibitor
- Adjust pH to 7.4 with 1 N NaOH
- Filter through 0.2 µm, make 0.3 ml aliquots and store at -20 °C for up to 6 months
- Culture medium (NB/B27 medium)
- Mix
485 ml of NBM
10 ml of B27
1.25 ml GlutaMAX
5 ml 100x penicillin/streptomycin (100 U/ml penicillin/100 μg/ml streptomycin final) - Filter through 0.2 μm filter, aliquot in 50 ml and store at 4 °C
- Artificial cerebrospinal fluid (1x)
125 mM NaCl
10 mM NaHCO3
25 mM D-glucose
2.5 mM KCl
2 mM CaCl2
1.25 mM NaH2PO4
1 mM MgCl2
10 mM HEPES - Intracellular solution (1x)
130 mM K-gluconate
10 mM HEPES
10 mM phosphocreatine
5 mM KCl
1 mM MgCl2
4 mM ATP-Mg
0.3 mM GTP
Acknowledgments
Jeremie Barral was supported by a Human Frontier Science Program long-term postdoctoral fellowship (LT000132/2012) and by the Bettencourt Schueller Foundation. Alex Reyes was supported by grants from the National Institutes of Health (DC005787-01A1). This protocol was adapted from procedures published in Barral and Reyes (2016).
References
- Anselmi, F., Banerjee, A. and Albeanu, D. F. (2015). Patterned photostimulation in the brain. In: Douglass, A. D. (Ed.). New Techniques in Systems Neuroscience. Springer 235-270.
- Barral, J. and Reyes, A. (2016). Synaptic scaling rule preserves excitatory-inhibitory balance and salient neuronal network dynamics. Nat Neurosci 19(12): 1690-1696.
- Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G. and Deisseroth, K. (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 8(9): 1263-1268.
- Brewer, G. J., Torricelli, J. R., Evege, E. K. and Price, P. J. (1993). Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J Neurosci Res 35(5): 567-576.
- Farah, N., Reutsky, I. and Shoham, S. (2007). Patterned optical activation of retinal ganglion cells. Conf Proc IEEE Eng Med Biol Soc 2007: 6368-6370.
- Guo, Z. V., Hart, A. C. and Ramanathan, S. (2009). Optical interrogation of neural circuits in Caenorhabditis elegans. Nat Methods 6(12): 891-896.
- Hilgenberg, L. G. and Smith, M. A. (2007). Preparation of dissociated mouse cortical neuron cultures. J Vis Exp(10): 562.
- Luo, J., Deng, Z. L., Luo, X., Tang, N., Song, W. X., Chen, J., Sharff, K. A., Luu, H. H., Haydon, R. C., Kinzler, K. W., Vogelstein, B. and He, T. C. (2007). A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc 2(5): 1236-1247.
- Nawrot, M. P., Schnepel, P., Aertsen, A. and Boucsein, C. (2009). Precisely timed signal transmission in neocortical networks with reliable intermediate-range projections. Front Neural Circuits 3: 1.
- Papagiakoumou, E., Anselmi, F., Begue, A., de Sars, V., Gluckstad, J., Isacoff, E. Y. and Emiliani, V. (2010). Scanless two-photon excitation of channelrhodopsin-2. Nat Methods 7(10): 848-854.
- Shoham, S., O’Connor, D. H., Sarkisov, D. V. and Wang, S. S. (2005). Rapid neurotransmitter uncaging in spatially defined patterns. Nat Methods 2(11): 837-843.
- Stirman, J. N., Crane, M. M., Husson, S. J., Gottschalk, A. and Lu, H. (2012). A multispectral optical illumination system with precise spatiotemporal control for the manipulation of optogenetic reagents. Nat Protoc 7(2): 207-220.
- Stirman, J. N., Crane, M. M., Husson, S. J., Wabnig, S., Schultheis, C., Gottschalk, A. and Lu, H. (2011). Real-time multimodal optical control of neurons and muscles in freely behaving Caenorhabditis elegans. Nat Methods 8(2): 153-158.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Barral, J. and Reyes, A. D. (2017). Optogenetic Stimulation and Recording of Primary Cultured Neurons with Spatiotemporal Control. Bio-protocol 7(12): e2335. DOI: 10.21769/BioProtoc.2335.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell signaling > Intracellular Signaling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.