- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Single-molecule Analysis of DNA Replication Dynamics in Budding Yeast and Human Cells by DNA Combing

Published: Vol 7, Iss 11, Jun 5, 2017 DOI: 10.21769/BioProtoc.2305 Views: 15242
Reviewed by: Emilie BesnardAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2012
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
The DNA combing method allows the analysis of DNA replication at the level of individual DNA molecules stretched along silane-coated glass coverslips. Before DNA extraction, ongoing DNA synthesis is labeled with halogenated analogues of thymidine. Replication tracks are visualized by immunofluorescence using specific antibodies. Unlike biochemical and NGS-based methods, DNA combing provides unique information on cell-to-cell variations in DNA replication profiles, including initiation and elongation. Finally, this assay can be used to monitor the effect of DNA lesions on fork progression, arrest and restart.
Keywords: ReplicationBackground
DNA synthesis is initiated at thousands of sites on eukaryotic chromosomes called replication origins. Origin activation follows a well-defined replication timing program that is controlled by checkpoint kinases and epigenetic modifications of chromatin (Prioleau and MacAlpine, 2016). Replication forks frequently stall during a normal S phase. Fork arrest is caused by multiple events, such as DNA lesions, tightly bound protein complexes, and transcription at highly expressed genes (Tourriere and Pasero 2007; Zeman and Cimprich, 2013). Eukaryotes have developed different strategies to deal with this replication stress, including repair mechanisms to restart arrested forks and activation of dormant replication origins to rescue terminally-arrested forks.
DNA combing is a method of choice to monitor different aspects of replication (fork speed, origin usage, fork restart, sister fork asymmetry). Unlike other DNA fiber methods such as DNA fiber spreading, the stretching, density and alignment of DNA molecules are highly reproducible and tightly controlled in the DNA combing method. Stretching is imposed by the force exerted by a receding air/water interface, independently of the length of DNA fibers (Bensimon et al., 1994; Michalet et al., 1997). Origin firing and progression of replication forks are followed after incorporation of thymidine analogs, such as 5-bromo-2’-deoxyuridine (BrdU), 5-iodo-2’-deoxyuridine (IdU) and 5-chloro-2’-deoxyuridine (CldU) in newly-synthesized DNA. This technique has been successfully used to monitor DNA replication dynamics in a variety of organisms, including bacteria, yeast, Drosophila, Xenopus and mammals.
Here, we provide detailed protocols to analyze newly synthesized DNA fibers in budding yeast and in human cells and to investigate various aspects of DNA replication in normal growth conditions and under replicative stress.
Materials and Reagents
- Common to human/yeast cells
- Tape
- 14 ml round-bottom polypropylene tubes (Corning, Falcon®, catalog number: 352059 )
- Tips 1 ml, 200 µl, 20 µl
- Silanized coverslips (Genomic Vision, catalog number: COV-001 ) purchased from Genomic Vision or prepared as described (Labit et al., 2008)
- 2 ml Teflon reservoir (Reservoir MCS Support [x 2]; from Genomic Vision)
- Whatman paper
- Microscope slides SuperFrost (VWR, catalog number: 630-1987 )
- Cyanoacrylate glue
- Diamond tip engraving pen (Sigma-Aldrich, catalog number: Z225568-1EA )
- Saran plastic film (Dominique Dutscher, catalog number: 090264 )
- Coplin Jar
- EDTA (Sigma-Aldrich, catalog number: E6758 )
- LMP agarose (Bio-Rad Laboratories, catalog number: 161-3111 )
- Plug mold (Bio-Rad Laboratories, catalog number: 170-3713 )
- Proteinase K (Sigma-Aldrich, catalog number: P6556 )
- 10x PBS (Sigma-Aldrich, catalog number: D1408 )
- YOYO-1 (Thermo Fisher Scientific, InvitrogenTM, catalog number: Y3601 )
- Sodium chloride (NaCl) (VWR, catalog number: 27810-295 )
- β-agarase (New England Biolabs, catalog number: M0392L )
- Sodium hydroxide (NaOH) (Merck, catalog number: 1.06462.1000 )
- BSA fraction V (Sigma-Aldrich, catalog number: A9647 )
- Prolong Gold Antifade reagent (Thermo Fisher Scientific, InvitrogenTM, catalog number: P36930 )
- BrdU (Sigma-Aldrich, catalog number: B5002 )
- IdU (Sigma-Aldrich, catalog number: I7125 )
- CldU (MP Biomedicals, catalog number: 0 2105478 )
- DMSO (Sigma-Aldrich, catalog number: D2650 )
- Hydroxyurea (Sigma-Aldrich, catalog number: H8627 )
- N-laurylsarcosine sodium salt (Sigma-Aldrich, catalog number: L9150 )
- MES hydrate (Sigma-Aldrich, catalog number: M2933 )
- MES sodium salt (Sigma-Aldrich, catalog number: M5057 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- Mouse anti-BrdU clone B44 IgG1 (BD, BD Biosciences, catalog number: 347580 )
- Rat anti-BrdU clone BU1/75 (Bio-Rad Laboratories, catalog number: OBT0030 )
- Mouse anti ssDNA (poly dT) IgG2a (EMD Millipore, catalog number: MAB3034 or MAB3868 )
- Goat anti-Rat Alexa 488 (Thermo Fisher Scientific, Invitrogen, catalog number: A-11006 )
- Goat anti-Mouse Alexa 546 (Thermo Fisher Scientific, Invitrogen, catalog number: A-11030 )
- Goat anti-Mouse IgG2a Alexa 647 (Thermo Fisher Scientific, Invitrogen, catalog number: A-21241 )
- Goat anti-Mouse IgG1 Alexa 546 (Thermo Fisher Scientific, Invitrogen, catalog number: A-21123 )
- LMP agarose (see Recipes)
- TE50 (see Recipes)
- TE 1x (see Recipes)
- 10x MES buffer pH 6 (see Recipes)
- 1 N NaOH (see Recipes)
- PBS/T (see Recipes)
- Antibodies (dilution in PBS/T) (see Recipes)
- Detection of CldU/IdU/ssDNA (see Recipes)
- Detection of BrdU/ssDNA (see Recipes)
- Tape
- S. cerevisiae specific reagents
- Yeast strain PP872 (genetic background: W303; genotype: MATa, ade2-1, trp1-1, can1-100, leu2-3,112, his3-11,15, ura3, GAL, psi+, RAD5, ura3::URA3-GPD-TK7) (Crabbe et al., 2010)
- Yeast strain HSV-TK + hENT1 (Viggiani and Aparicio, 2006)
- Bacto peptone (BD, BactoTM, catalog number: 211677 )
- Adenine (Sigma-Aldrich, catalog number: A8626 )
- Yeast extract (Sigma-Aldrich, catalog number: 70161 )
- Glucose (VWR, catalog number: 101175P )
- Alpha factor (custom peptide synthesis, Sequence: WHWLQLKPGQPMY)
- Pronase (EMD Millipore, catalog number: 53702-50KU )
- Sodium azide (NaN3) (Sigma-Aldrich, catalog number: 71289 )
- Tris-HCl (Sigma Aldrich, catalog number: RES3098T )
- Citric acid monohydrate (C6H8O7·H2O) (Sigma-Aldrich, catalog number: C1909 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S3264 )
- Monopotassium phosphate (KH2PO4) (VWR, catalog number: 26936.293 )
- Dibasic potassium phosphate (K2HPO4) (VWR, catalog number: 26930.293 )
- Enzyme powder Zymolyase 20T (MP Biomedicals, catalog number: 0 8320921 )
- YPAD (see recipes)
- Zymolyase buffer (see Recipes)
- 1 M potassium phosphate buffer pH 7.0 (see Recipes)
- 0.1 M citrate phosphate buffer pH 5.6 (see Recipes)
- BrdU stock solution (see Recipes)
- Proteinase K buffer (see Recipes)
- Yeast strain PP872 (genetic background: W303; genotype: MATa, ade2-1, trp1-1, can1-100, leu2-3,112, his3-11,15, ura3, GAL, psi+, RAD5, ura3::URA3-GPD-TK7) (Crabbe et al., 2010)
- Human cells specific reagents
Equipment
- Centrifuge (Eppendorf, model: 5810 R )
- Microcentrifuge (Eppendorf, model: MiniSpin® )
- Thermoblock (thermomixer comfort Eppendorf with 2 ml; 15 ml and 50 ml block)
- Cell counter (CASY® Modell TT - Cell Counter, OLS for yeast cells or Malassez hemocytometer [VWR, catalog number: 631-0975 ] for human cells)
- Pasteur pipette Rubber bulb (Dominique Dutscher, catalog number: 042250 )
- Microwave
- Roller mixer (Cole-Parmer, Stuart, model: SRT9D )
- Leica DM6000B microscope (Leica Microsystems, model: DM6000B )
- CoolSNAP HQ CCD camera (Photometrics, model: CoolSNAP HQ CCD )
- P1000, P200, P20 pipetman
- DNA combing device (Genomic Vision, catalog number: MCS-001 ). Can also be assembled as described (Gallo et al., 2016; Kaykov et al., 2016; Norio and Schildkraut, 2001)
- Metal coverslip holder
- Humid chamber (StainTray slide staining system) (Sigma-Aldrich, catalog number: Z670146-1EA )
- Hybridization oven (Shake ‘n’ StackTM Hybridization Ovens) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 6240TS )
- Epifluorescence microscope Zeiss Axio Imager Z2 (Zeiss, model: Axio Imager Z2 ) with a camera Hamamatsu ORCA-Flash4.0 LT, Cmos sensor of 6.5 µm with an objective Zeiss 40x PL APO 1.4 oil and with the filters GFP HE: BP470/40 FT495 BP525/50; Texas Red: BP560/40 FT585 BP630/75; CY5: BP 640/30 FT660 BP690/50)
- Microscope (Nikon Instruments, model: YS100 )
Software
- MetaMorph (Molecular Devices) or open source software such as ImageJ (http://rsbweb.nih.gov/ij/) with bio-formats plugins
- IDeFIx Montpellier (IGMM, CNRS) or software developed by Genomic Vision
- Prism 7.0 (GraphPad) or open source software such as R (https://www.r-project.org/)
Procedure
- Saccharomyces cerevisiae
S. cerevisiae cells are unable to incorporate BrdU into DNA because they lack the nucleotide salvage pathway that enables uptake of extracellular thymidine or its analogs. To circumvent this problem, strains for molecular combing have been engineered to ectopically express the Herpes simplex virus thymidine kinase (HSV-TK) under the control of the constitutive GPD promoter, inserted at the URA3 locus on chromosome V (strain PP872; genetic background: W303; genotype: MATa, ade2-1, trp1-1, can1-100, leu2-3,112, his3-11,15, ura3, GAL, psi+, RAD5, ura3::URA3-GPD-TK7) (Lengronne et al., 2001; Viggiani and Aparicio, 2006). Cells bearing one integrated copy of the HSV-TK + hENT1 vector (human equilibrative nucleoside transporter 1) (Viggiani and Aparicio, 2006) show comparable levels of BrdU incorporation to cells with seven copies of HSV-TK alone (Bianco et al., 2012).
Note: DNA molecular combing procedure doesn’t differ for fission yeast excepted for growth and labelling conditions (Kaykov et al., 2016).- BrdU labeling of synchronized HU-arrested cells and preparation of DNA genomic plugs
- Grow cells overnight at 25 °C to a density of 5 x 106 cells/ml in 100 ml of YPAD.
- Add α-factor (2 μg/ml) at T0 in order to synchronized cells in G1. A second (4 μg/ml) and a third dose (2 μg/ml) of α-factor are added part way at respectively T1h and T2h to ensure that cells do not escape the G1 arrest.
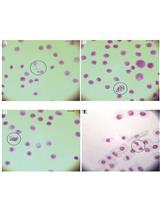
Note: The length of G1 synchronisation varies from 2.5 to 3 h depending on the doubling time of yeast strains. Make sure that cells are in G1 (unbudded cells) before the release into S phase (Figure 1).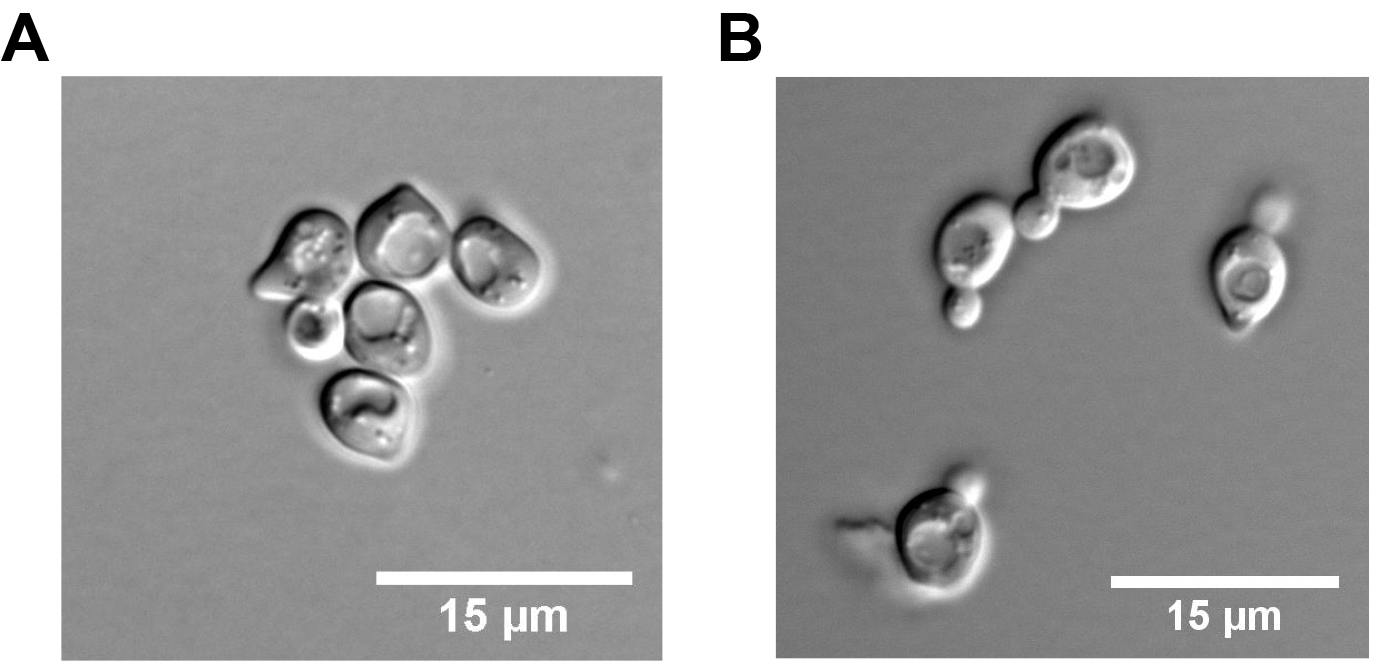
Figure 1. Yeast cell morphology in G1 and early S phase. A. G1 synchronised yeast cells; B. Yeast cells in S phase + HU after release from G1 for 90 min. - Add (i) BrdU to a final concentration of 400 μg/ml (or 40 μg/ml if cells express the nucleotide transporter hENT1), and (ii) Hydroxyurea (HU, 200 mM) at least 15 min before releasing cells into S phase.
Note: Elongation from early-firing origins is inhibited with the addition of HU in order to restrict the length of newly replicated regions to 10 kb. - Release cells from α-factor arrest into S phase by addition of 50 μg/ml Pronase (degrades alpha factor peptide in medium). Adjust the pH of the medium to 7.0 with potassium phosphate buffer.
- After 90 min, collect cells by centrifugation at 4 °C; 1,096 x g in a 50 ml tube containing cold 0.1% NaN3 (sodium azide).
Note: Sodium azide inhibits cytochrome oxidase (last enzyme in the respiratory electron transport chain of mitochondria or bacteria) by binding irreversibly to the heme cofactor and stops all cell metabolism, including DNA replication. - Resuspend the cells in one volume of cold TE50, 0.1% NaN3 and keep them on ice. Check cells under a microscope (Nikon YS100). More than 90% of the cells should display small buds, which are indicative of entry into S phase (Figure 1).
- Grow cells overnight at 25 °C to a density of 5 x 106 cells/ml in 100 ml of YPAD.
- Preparation of genomic DNA plugs
Genomic DNA is prepared in low melting point (LMP) agarose plugs to prevent mechanical shearing.- Freshly prepare molten 2% LMP agarose in bi-distilled water and Zymolyase buffer. Keep at 42 °C in thermomixer until use.
- Determine cell concentration with a cell counter.
- Wash cells once with NZ buffer.
- Resuspend cells in prewarmed Zymolyase buffer (42 °C) to a final concentration of 4 x 108 cells/ml and carefully mixed with an equal volume of molten 2% LMP agarose (42 °C).
Note: The Zymolyase will digest the cell wall. - Transfer the cellular suspension into a plug mold sealed with tape to generate 90 μl plugs (Figure 2) containing 2 x 107 cells or approximately 350 ng of genomic DNA per plug at room temperature.
Note: Prepare at least 4 plugs per sample. - Cover molds with a plastic film (Figure 2) and incubate for 30 min at 37 °C.

Figure 2. Pictures of a plug mold. A and B. Top and bottom views; C. Plug mold sealed with a tape and covered with a saran plastic film. - Place agarose plugs at 4 °C for 10 min to let the agarose solidify.
- Transfer plugs into 14 ml round-bottom polypropylene tubes using a Pasteur pipette rubber bulb and incubate them at 37 °C in Proteinase K buffer (2 ml for 4 plugs) for 1 h under gentle agitation. Transfer plugs into fresh Proteinase K buffer and incubate for 24 h at 37 °C. Repeat once for an additional 24 h.
Note: Incubation with PK buffer is critical to degrade proteins that perturb DNA fiber stretching. - Wash plugs for two days in TE50, 100 mM NaCl on a roller mixer at room temperature. Change the buffer from time to time.
- Keep plugs at 4 °C in TE50 buffer until use (stable for months). Note that DNA plugs are translucent and fragile.
- Freshly prepare molten 2% LMP agarose in bi-distilled water and Zymolyase buffer. Keep at 42 °C in thermomixer until use.
- BrdU labeling of synchronized HU-arrested cells and preparation of DNA genomic plugs
- Human cells
Note: This protocol is available for any kind of mammalian cells.- BrdU labelling and preparation of DNA genomic plugs
Unlike budding yeast, human cells cannot be easily synchronized and experiments are usually performed with asynchronous cultures. Under these conditions, it is critical to use a combination of two modified nucleotides (IdU and CldU) to determine the polarity of replication forks and the position of initiation sites (Figure 3). (Conti et al., 2007)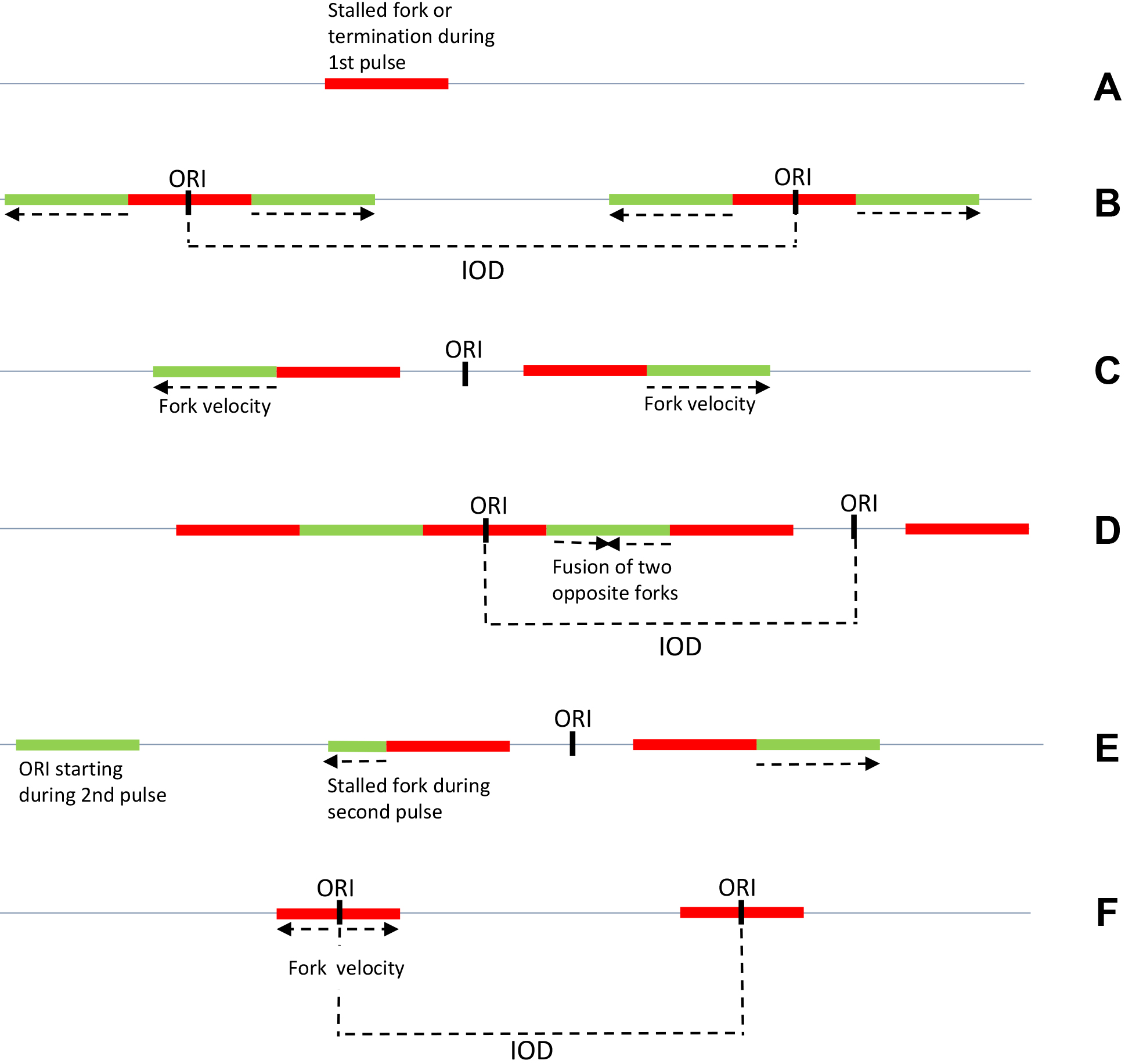
Figure 3. Schematic patterns obtained by DNA combing after IdU/CldU pulses in asynchronous mammalian cells. To determine fork velocity, green tracks adjacent to red tracks are measured in B, C, E. Stalled forks are represented in A and E and interorigin distances (IOD) in B and D. ORI: origin of replication. Arrows indicate fork direction. F. Schematic pattern obtained by DNA combing after BrdU pulse in synchronized budding yeast cells.
- Grow cells to a confluence of about 70% in 6-well plates. At the time of the experiment each well need to contain a minimum of 1 x 105 cells.
- Label cells by adding IdU (20 μM final) directly in 2 ml medium.
- Incubate cells at 37 °C for at least 10 min.
- Replace medium with 2 ml of prewarmed medium containing 200 μM CldU. Incubate cells at 37 °C for 20 min.
Note: To avoid thermal shock and other perturbations, add nucleotides directly to growth medium (first pulse). Use prewarmed medium and a much higher concentration for the second pulse to avoid washes.
The range of IdU/CldU staining time should be adjusted for each cell line from 10 to 30 min depending on S phase duration. - Rinse cells with PBS and trypsinize cells as usual, adjust trypsinization time to cell type. For instance, incubate cells for 3-5 min in 0.05% trypsin, 1 mM EDTA.
- Collect cells, place them on ice and spin for 5 min at 1,000 x g at 4 °C. Keep the cells on ice until use (to stop DNA synthesis).
- Grow cells to a confluence of about 70% in 6-well plates. At the time of the experiment each well need to contain a minimum of 1 x 105 cells.
- Preparation of genomic DNA plugs
- Resuspend cells in 5 ml cold PBS, spin for 5 min at 1,000 x g at 4 °C and resuspend in 1 ml cold PBS.
- Count cells and resuspend at 8 x 105 to 2 x 106 cells/ml in cold PBS, depending on cell size.
- Prepare a solution of 2% LMP agarose in PBS and keep at 42 °C until use.
- Add an equal volume of 2% LMP agarose to cells. To prepare 4 plugs, add 200 μl of 2% LMP agarose solution to 200 μl of prewarmed cells solution.
- Mix very gently with a P1000 pipetman (optional: cut tip).
- Transfer 90 μl per plug into a plug mold sealed with tape.
- Let plugs solidify for 25 min at room temperature and for 10 min at 4 °C.
- Use a Pasteur pipette rubber bulb to eject plugs into 14 ml round-bottom tubes containing 0.5 ml of Proteinase K buffer per plug. Mix gently on roller mixer and change the PK buffer after 1 h.
- Incubate for 48 h at 37 °C. Change the PK solution twice more during this incubation. Mix gently on a roller mixer.
Note: Incubation with PK buffer is critical to degrade proteins that perturb DNA fiber stretching. - Gently remove the solution by blocking the agarose plug with a cell scraper. Plugs are transparent and difficult to manipulate after PK treatment.
- Wash plugs several times for two days in TE50, 100 mM NaCl on the roller mixer at room temperature.
- Store at 4 °C in TE50 until use. Genomic DNA is stable for months at 4 °C in TE50 buffer.
- BrdU labelling and preparation of DNA genomic plugs
- Procedures common to yeast and human cells
- Melting of agarose plugs containing genomic DNA
- Transfer a plug in a 12 ml round-bottom tube. Plugs are washed 3 times for 1 h in 1x TE pH 7.5, 100 mM NaCl.
- After removing of the last wash, add 100 μl 1x TE and 1.5 μl YOYO-1 to stain genomic DNA. Keep in the dark for 30 min at room temperature.
Note: YOYO staining is lost after DNA denaturation but is used to check combing before detection. - Wash 4 times for 5 min with 10 ml 1x TE with gentle shaking.
- Wash once for 5 min with 2 ml of 50 mM MES pH 6, 100 mM NaCl.
- Replace with 3 ml of 50 mM MES pH 6, 100 mM NaCl.
Note: Increase the volume of MES if fiber density is too high after combing. The pH of MES is critical to obtain a correct density and stretching of DNA fibers. - Incubate for 15-30 min for human cells and 45 min for yeast cells at 67 °C in a heating block.
Note: From now on, manipulate samples with extreme care to avoid DNA shearing. - Check the DNA solution. If the shape of the plug is still visible, put it back at 67 °C until complete melting.
- Let the solution cool down to 42 °C before adding β-agarase (3 U per plug).
- Incubate overnight at 42 °C.
- Visually inspect the DNA solution. If you still see agarose aggregates; add β-agarase (1 U per plug) for 2 more hours.
- Incubate at 65 °C for 10 min and store at room temperature in dim light until use.
- DNA combing
DNA combing is performed on silanized coverslips as described (Bensimon et al., 1994; Lengronne et al., 2001; Michalet et al., 1997). The quality of silanized coverslips is critical for the subsequent analysis of replication profiles. The whole procedure using the genomic vision device can be visualized on this video: https://youtu.be/LjjpYby3YIk.- Carefully pour the DNA solution into a 2 ml Teflon reservoir. Save the rest for further use.
Note: Genomics Vision sells disposable 2 ml plastic reservoirs to melt plug directly in the reservoir and avoid manipulation of the DNA solution. - Insert a silanized coverslip into the DNA solution and incubate for 5 min at room temperature.
- Using a DNA combing device, remove carefully the coverslip from the reservoir at a constant speed of 300 μm/sec. Repeat with another coverslip as many times as needed (2 slides per sample is usually sufficient).
- At this stage, DNA is stretched on both sides of the coverslip. Visualize DNA fibers on one side under the microscope (Nikon YS100) using a 40x objective and a FITC filter block. To this aim, fix with tape the coverslip to a metal coverslip holder, put a drop of immersion oil directly on the coverslip to visualize DNA fibers.
- Place the coverslip on a sheet of Whatman paper to soak up oil and bake for 2 h at 60 °C to crosslink DNA to coverslips.
- Stick the oiled side of coverslips on glass slides with cyanoacrylate glue. Label slides with a diamond tip engraving pen and store at -20 °C until use.
Note: If needed, some steps presented in A2/B2 and C1/C2 sections can be modified to increase the length of stretched DNA molecules as described (Kaykov et al., 2016). If the density of fibers is too low, the DNA solution can be kept for a few days at 4 °C to increase DNA resuspension in MES buffer before combing.
- Carefully pour the DNA solution into a 2 ml Teflon reservoir. Save the rest for further use.
- Immunodetection
Specific combinations of primary and secondary antibodies are used to detect BrdU or CldU/IdU and DNA fibers simultaneously. - Dehydrate slides for 5 min in successive baths of 70%, 90% and 100% ethanol in Coplin jar. Let air dry the slides.
- Incubate in Coplin jar for 25 min in freshly prepared 1 N NaOH to denature the DNA duplex.
Note: Prolonged incubation in NaOH degrades the silane layer and should be avoided. - Wash extensively with PBS pH 7.4 to neutralize NaOH (5 washes of 1 min each).
- Saturate slides for 15 min in PBS/T containing 1% BSA.
- Add 20 μl of PBS/T containing primary antibodies and cover with a coverslip. Incubate for 45 min at 37 °C in a humid chamber.
- Wash 5 min x 2 times with PBS/T in Coplin jar.
Note: Dip slide in a Coplin jar containing PBS/T in order to remove the upper coverslip without damaging the DNA fibers. - Detect with secondary antibodies (30 min at 37 °C in a humid chamber).
- Wash 5 min x 3 times with PBS/T in Coplin Jar.
- Air dry slides and mount by adding 5 μl Prolong Gold Antifade reagent on coverslip covered with a second coverslip. Let mounting reagent polymerize for at least 2 h at room temperature (preferably overnight and protect from light) before proceeding with microscopy. Mounted coverslips are stable for months at -20 °C.
- Image acquisition and data analysis
- Image acquisition is performed with a 40x objective on a motorized microscope equipped with a camera and controlled with Zeiss ZEN Pro 2 software or equivalent (Figure 4). The conversion factor (CF) from pixel to bp (CF = P/M x S) depends on the pixel size of the CCD camera (P in μm), on the magnification of the objective (M) and on the stretching of DNA fibers (S, 2 kb/µm for DNA combing). DNA molecules of known length, such as concatemers of bacteriophage lambda DNA, can be used as size standards.
- BrdU tracks and DNA fibers can be measured manually with MetaMorph (Molecular Devices) or open source software such as ImageJ (http://rsbweb.nih.gov/ij/). Data are transferred to an Excel spreadsheet for analysis. DNA fiber identification and length measurements can be automated with software like IDeFIx, developed by the DNA combing facility of Montpellier (IGMM, CNRS) or software developed by Genomic Vision. Critical issues regarding potential biases and limitations of DNA fiber analysis have been extensively discussed elsewhere (Techer et al., 2013; Tuduri et al., 2010).
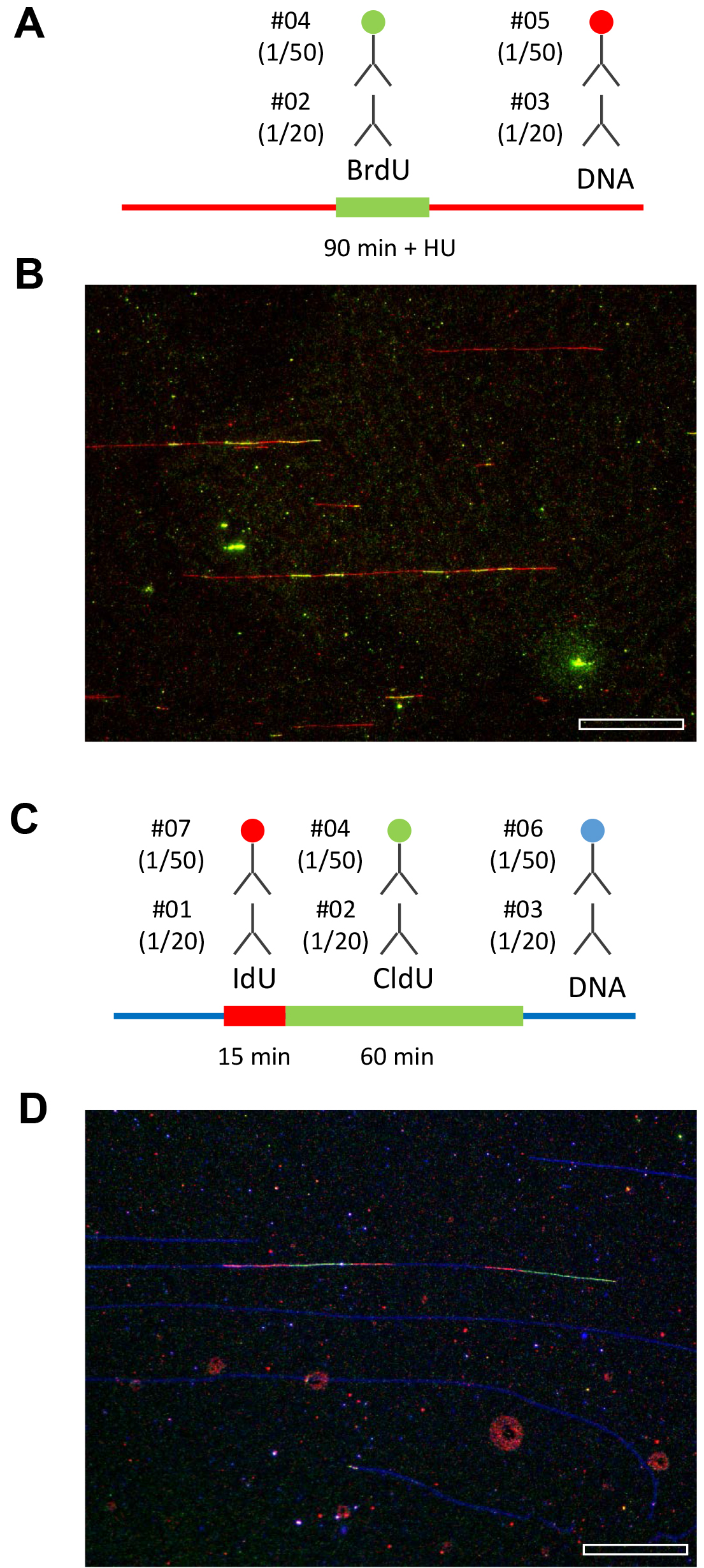
Figure 4. Representative examples of replication patterns observed by DNA combing. A and B. Yeast cells were released from G1 in the presence of 200 mM HU and labeled with BrdU for 90 min. DNA fibers were purified and stretched by DNA combing. BrdU and ssDNA were detected as indicated. A representative field of view is shown. Bar is 50 kb. C and D. Human cells were labeled for 15 min with IdU and 60 min with CldU. The combination of antibodies used to detect IdU, CldU and ssDNA is indicated. A representative field of view is shown. Bar is 50 kb. Image acquisition is performed with a motorized Leica DM6000B microscope equipped with a CoolSNAP HQ CCD camera (6.45 µm/pixel) and a 40x oil immersion objective. With this setup and a DNA stretching of 2 kb/µm, one pixel corresponds to (6.45/40) x 2 = 323 bp.
Data analysis
- Statistical analysis of BrdU track length and inter-origin distances is performed with Prism 7.0 (GraphPad) or R Statistical software. Since BrdU track lengths and inter-origin distances do not display a Gaussian distribution, statistical significance of differences between samples is tested using nonparametric tests such as the Mann-Whitney rank sum test.
- To determine fork speed, the median length of CldU tracks flanked with IdU tracks on one side (single fork) is divided by the duration of the second pulse (usually 20 min). CldU tracks alone and CldU tracks surrounded with two IdU tracks are not taken into account. CldU and IdU can also be used to measure inter-origin distances (IODs), global fork densities (Bialic et al., 2015) and to monitor replication fork arrest/restart (Techer et al., 2013; Tuduri et al., 2009) (Figure 3).
- IdU and CldU pulses can be used to visualize the progression of sister replication forks. In normal growth conditions, these signals are normally symmetrical as sister forks progress with the same speed. When asymmetrical patterns are detected this is indicative of increased replication fork pausing or stalling (Tuduri et al., 2009). This increased rate of fork stalling can be expressed as the ratio of the longest to the shortest CldU tracks, or represented graphically as a scatter plot of the distance covered by the two sister forks during the CldU pulse. Large ratio or dispersed scatter plots are indicative of increased fork arrest.
- Another useful application of IdU/CldU double labeling is the analysis of DNA replication recovery after a genotoxic insult (Sidorova et al., 2013; Tourriere et al., 2005). In this assay, cells are exposed to genotoxic agents or replication inhibitors such as hydroxyurea (HU), methyl methanesulfonate (MMS) or camptothecin (CPT) in the presence of IdU and are released in fresh medium in the presence of CldU. This approach can be used to monitor cells ability to restart stalled forks, to activate late or dormant origins and to complete DNA replication after release from the drug.
- It has been recently reported that stalled forks can be processed by nucleases. Resection of nascent DNA at forks arrested by genotoxic agents such as HU or CPT can be visualized after IdU and CldU pulses as described (Ray Chaudhuri et al., 2016; Schlacher et al., 2011).
Recipes
- Yeast cells
- YPAD
1% yeast extract
2% Bacto peptone
0.005% adenine
2% glucose
Bi-distilled water - BrdU
10 mg/ml stock solution in Milli-Q H2O (freshly prepared) - Zymolyase buffer
50 mM citrate phosphate buffer, pH 5.6
50 mM EDTA, pH 8.0
1.2 M sorbitol
1 mg/ml Zymolyase 20T (use enzyme powder) - NZ (no Zymolyase) buffer
50 mM citrate phosphate buffer, pH 5.6
50 mM EDTA, pH 8.0
1.2 M sorbitol - 1 M potassium phosphate buffer pH 7.0; 100 ml
61.5 ml KH2PO4, 2 M (M.W. = 136)
38.5 ml K2HPO4, 2 M (M.W. = 174) - 0.1 M citrate phosphate buffer pH 5.6; 100 ml
58 ml of 0.2 M Na2HPO4 (dibasic; M.W. = 142)
42 ml of 0.1 M citric acid (C6H8O7·H2O; M.W. = 210) - Proteinase K buffer
125 mM EDTA pH 9.5
1% N-laurylsarcosine
1 mg/ml Proteinase K
- YPAD
- Human cells
- BrdU: 25 mM stock solution in PBS, 10% DMSO. Store at -20 °C
- IdU: 25 mM stock solution in PBS, 10% DMSO. Store at -20 °C
- CldU: 200 mM stock solution in PBS, 10% DMSO. Store at -20 °C
- Proteinase K buffer
10 mM Tris pH 7.5
100 mM EDTA pH 8.0
1% N-laurylsarcosine
1 mg/ml Proteinase K
- BrdU: 25 mM stock solution in PBS, 10% DMSO. Store at -20 °C
- Common to human/yeast cells
- LMP agarose, freshly prepared, check volume: 2% in water for yeast cells or in PBS for human cells. LMP can be melted in a microwave at a very low power; please make sure to avoid any boiling of the solution
- TE50
10 mM Tris-HCl, pH 7.0
50 mM EDTA, pH 8.0 - 1x TE
10 mM Tris-HCl, pH 7.0
1 mM EDTA, pH 8.0 - 10x MES buffer pH 6
70 ml of 500 mM MES hydrate
30 ml of 500 mM MES sodium salt
Adjust to pH 6 with 500 mM MES sodium salt - 1 N NaOH (freshly prepared)
- PBS/T
PBS pH 7.4
0.1% Triton X-100 - Antibodies (dilution in PBS/T)
# 01 Mouse anti-BrdU clone B44 IgG1
# 02 Rat anti-BrdU clone BU1/75
# 03 Mouse anti ssDNA (poly dT) IgG2a
# 04 Goat anti-Rat Alexa 488
# 05 Goat anti-Mouse Alexa 546
# 06 Goat anti-Mouse IgG2a Alexa 647
# 07 Goat anti-Mouse IgG1 Alexa 546 - Detection of CldU/IdU/ssDNA
Primary antibodies:
CldU: #01 (1/20)
IdU: #02 (1/20)
ssDNA: #03 (1/50)
Secondary antibodies:
#07 (1/50)
#04 (1/50)
#06 (1/50) - Detection of BrdU/ssDNA
Primary antibodies:
BrdU: #02 (1/20)
ssDNA: #03 (1/50)
Secondary antibodies:
#04 (1/50)
#05 (1/50)
- LMP agarose, freshly prepared, check volume: 2% in water for yeast cells or in PBS for human cells. LMP can be melted in a microwave at a very low power; please make sure to avoid any boiling of the solution
Acknowledgments
We thank the members of the Pasero laboratory for their contribution to the optimization of this protocol, which was adapted from protocols of several other groups, including the Bensimon, Schwob, Debatisse and Nurse laboratories. We thank the Montpellier RIO Imaging facility for support and the DNA combing facility of Montpellier for providing silanized coverslips. This work was supported by grants from Agence Nationale pour la Recherche (ANR), Institut National du Cancer (INCa), Ligue contre le Cancer (Equipe labellisée LIGUE, 2017) and the MSDAvenir fund.
References
- Bensimon, A., Simon, A., Chiffaudel, A., Croquette, V., Heslot, F. and Bensimon, D. (1994). Alignment and sensitive detection of DNA by a moving interface. Science 265(5181): 2096-2098.
- Bialic, M., Coulon, V., Drac, M., Gostan, T. and Schwob, E. (2015). Analyzing the dynamics of DNA replication in Mammalian cells using DNA combing. Methods Mol Biol 1300: 67-78.
- Bianco, J. N., Poli, J., Saksouk, J., Bacal, J., Silva, M. J., Yoshida, K., Lin, Y. L., Tourriere, H., Lengronne, A. and Pasero, P. (2012). Analysis of DNA replication profiles in budding yeast and mammalian cells using DNA combing. Methods 57(2): 149-157.
- Conti, C., Sacca, B., Herrick, J., Lalou, C., Pommier, Y. and Bensimon, A. (2007). Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol Biol Cell 18(8): 3059-3067.
- Crabbe, L., Thomas, A., Pantesco, V., De Vos, J., Pasero, P. and Lengronne, A. (2010). Analysis of replication profiles reveals key role of RFC-Ctf18 in yeast replication stress response. Nat Struct Mol Biol 17(11): 1391-1397.
- Gallo, D., Wang, G., Yip, C. M. and Brown, G. W. (2016). Single-molecule analysis of replicating yeast chromosomes. Cold Spring Harb Protoc 2016(2): pdb top077784.
- Kaykov, A., Taillefumier, T., Bensimon, A. and Nurse, P. (2016). Molecular combing of single DNA molecules on the 10 megabase scale. Sci Rep 6: 19636.
- Labit, H., Goldar, A., Guilbaud, G., Douarche, C., Hyrien, O. and Marheineke, K. (2008). A simple and optimized method of producing silanized surfaces for FISH and replication mapping on combed DNA fibers. Biotechniques 45(6): 649-652, 654, 656-648.
- Lengronne, A., Pasero, P., Bensimon, A. and Schwob, E. (2001). Monitoring S phase progression globally and locally using BrdU incorporation in TK+ yeast strains. Nucleic Acids Res 29(7): 1433-1442.
- Michalet, X., Ekong, R., Fougerousse, F., Rousseaux, S., Schurra, C., Hornigold, N., van Slegtenhorst, M., Wolfe, J., Povey, S., Beckmann, J. S. and Bensimon, A. (1997). Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science 277(5331): 1518-1523.
- Norio, P. and Schildkraut, C. L. (2001). Visualization of DNA replication on individual Epstein-Barr virus episomes. Science 294(5550): 2361-2364.
- Prioleau, M. N. and MacAlpine, D. M. (2016). DNA replication origins-where do we begin? Genes Dev 30(15): 1683-1697.
- Ray Chaudhuri, A., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J. E., Wong, N., Lafarga, V., Calvo, J. A., Panzarino, N. J., John, S., Day, A., Crespo, A. V., Shen, B., Starnes, L. M., de Ruiter, J. R., Daniel, J. A., Konstantinopoulos, P. A., Cortez, D., Cantor, S. B., Fernandez-Capetillo, O., Ge, K., Jonkers, J., Rottenberg, S., Sharan, S. K. and Nussenzweig, A. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535(7612): 382-387.
- Schlacher, K., Christ, N., Siaud, N., Egashira, A., Wu, H. and Jasin, M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145(4): 529-542.
- Sidorova, J. M., Kehrli, K., Mao, F. and Monnat, R., Jr. (2013). Distinct functions of human RECQ helicases WRN and BLM in replication fork recovery and progression after hydroxyurea-induced stalling. DNA Repair (Amst) 12(2): 128-139.
- Techer, H., Koundrioukoff, S., Azar, D., Wilhelm, T., Carignon, S., Brison, O., Debatisse, M. and Le Tallec, B. (2013). Replication dynamics: biases and robustness of DNA fiber analysis. J Mol Biol 425(23): 4845-4855.
- Tourriere, H. and Pasero, P. (2007). Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst) 6(7): 900-913.
- Tourriere, H., Versini, G., Cordon-Preciado, V., Alabert, C. and Pasero, P. (2005). Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol Cell 19(5): 699-706.
- Tuduri, S., Crabbe, L., Conti, C., Tourriere, H., Holtgreve-Grez, H., Jauch, A., Pantesco, V., De Vos, J., Thomas, A., Theillet, C., Pommier, Y., Tazi, J., Coquelle, A. and Pasero, P. (2009). Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 11(11): 1315-1324.
- Tuduri, S., Tourriere, H. and Pasero, P. (2010). Defining replication origin efficiency using DNA fiber assays. Chromosome Res 18(1): 91-102.
- Viggiani, C. J. and Aparicio, O. M. (2006). New vectors for simplified construction of BrdU-Incorporating strains of Saccharomyces cerevisiae. Yeast 23(14-15): 1045-1051.
- Zeman, M. K. and Cimprich, K. A. (2013). Causes and consequences of replication stress. Nat Cell Biol 16: 2-9.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tourrière, H., Saksouk, J., Lengronne, A. and Pasero, P. (2017). Single-molecule Analysis of DNA Replication Dynamics in Budding Yeast and Human Cells by DNA Combing. Bio-protocol 7(11): e2305. DOI: 10.21769/BioProtoc.2305.
Category
Cancer Biology > General technique > Biochemical assays > DNA replication
Biochemistry > DNA > Single-molecule Activity > Imaging
Molecular Biology > DNA > DNA labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.