- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation, Culturing, and Differentiation of Primary Myoblasts from Skeletal Muscle of Adult Mice

Published: Vol 7, Iss 9, May 5, 2017 DOI: 10.21769/BioProtoc.2248 Views: 28690
Reviewed by: Antoine de MorreeXiaoyi ZhengRakesh Bam
Original research article
The authors used this protocol in:

Dec 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
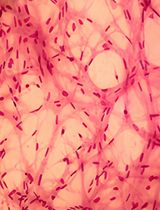
Abstract
Myogenesis is a multi-step process that leads to the formation of skeletal muscle during embryonic development and repair of injured myofibers. In this process, myoblasts are the main effector cell type which fuse with each other or to injured myofibers leading to the formation of new myofibers or regeneration of skeletal muscle in adults. Many steps of myogenesis can be recapitulated through in vitro differentiation of myoblasts into myotubes. Most laboratories use immortalized myogenic cells lines that also differentiate into myotubes. Although these cell lines have been found quite useful to delineating the regulatory mechanisms of myogenesis, they often show a great degree of variability depending on the origin of the cells and culture conditions. Primary myoblasts have been suggested as the most physiologically relevant model for studying myogenesis in vitro. However, due to their low abundance in adult skeletal muscle, isolation of primary myoblasts is technically challenging. In this article, we describe an improved protocol for the isolation of primary myoblasts from adult skeletal muscle of mice. We also describe methods for their culturing and differentiation into myotubes.
Keywords: MyoblastBackground
Myogenesis is a complex and highly orchestrated process that involves the determination of multipotential mesodermal cells to give rise to myoblasts, exit of myoblasts from the cell cycle, and their eventual differentiation into skeletal muscle fibers. Myogenesis is regulated by the sequential expression of myogenic regulatory factors (MRFs), a group of basic helix-loop-helix transcription factors that include Myf-5, MyoD, myogenin, and MRF4. Myf-5 and MyoD are the primary MRFs required for the formation, proliferation, and survival of myoblasts, whereas other MRFs such as myogenin and MRF-4 act late during myogenesis, activating gene expression of contractile proteins and other structural and metabolic proteins (Buckingham et al., 2003; Bentzinger et al., 2012).
Myogenesis is also regulated by a number of transcription factors and several noncoding RNAs, which act at specific steps including commitment of progenitor (satellite) cells to myogenic lineage and myoblast proliferation, differentiation, and fusion (Yin et al., 2013; Simionescu-Bankston and Kumar, 2016). Initial experiments for studying the role of various regulatory proteins in myogenesis are performed using cultured myoblasts. There are several myoblastic cell lines (e.g., C2C12, L6, BC3H1, and MM14) that differentiate into myotubes upon incubation in differentiation medium. These cell lines have also been used to establish myotube cultures to investigate the effects of various molecules on myotube growth and atrophy. However, there is often some degree of variability in results potentially due to the origin of cells, culture conditions, and passage number. The use of primary myoblasts is highly recommended because they are devoid of the side-effects characteristic of the immortalization process and their physiological relevance to the living organisms. Primary myoblasts can be isolated from the skeletal muscle of neonatal or adult mice. However, the process of isolation of myoblasts from neonatal muscle is more complex because it also requires Percoll density gradient centrifugation (Dogra et al., 2006). Some investigators also use fluorescence-activated cell sorting (FACS) approach to isolate primary myoblasts from digested muscle tissues especially to study regulation of quiescence and activation of these cells. However, FACS sorting is an expensive approach which requires several negative and positive selection antibodies and a cell sorter machine. Moreover, the yield of myoblasts is generally low and there are always chances of contamination during isolation of purified myoblasts by FACS technique. In our laboratory, we have adapted and standardized a previously published protocol (Rando and Blau, 1994) for the isolation of myoblasts from skeletal muscle of adult mice. This protocol is highly efficient for the generation of a large amount of purified myoblasts from skeletal muscle of adult mice (Ogura et al., 2015; Hindi and Kumar, 2016). The purity of the myoblasts can be assayed by immunostaining of the cells for Pax7 and MyoD proteins which are expressed in undifferentiated myoblasts. Moreover, primary myoblasts isolated using this protocol efficiently differentiate into multinucleated myotubes on incubation in differentiation medium and myotubes can be readily visualized by phase contrast microscopy or after immunostaining for myosin heavy chain (MyHC), a protein expressed in differentiated muscle cells (Hindi et al., 2014; Bohnert et al., 2016). Finally, like myogenic cell lines, the purified primary myoblasts can be stored in liquid nitrogen or -80 °C for unlimited time and can be regrown whenever required.
Materials and Reagents
- Sterilization pouches (Fisher Scientific, catalog number: 01-812-51 )
- 100 x 20 mm-Petri dishes (Corning, catalog number: 430167 )
- 6-well plates (Corning, Falcon®, catalog number: 353046 )
- 1.5 ml Eppendorf tubes (USA Scientific, catalog number: 1615-5510 )
- 0.22 μm filter (EMD Millipore, catalog number: SLGP033RS )
- 15 ml sterile tubes (VWR, catalog number: 89004-368 )
- 1 ml pipette tip
- Parafilm
- 10 ml serological pipette (Santa Cruz Biotechnology, catalog number: sc-200281 )
- 70 µm strainer (Fisher Scientific, catalog number: 22-363-548 )
- 50 ml sterile tubes (VWR, catalog number: 89004-364 )
- 30 µm filters (Milteny Biotech, catalog number: 130-041-407 )
- 0.45 μm filter (EMD Millipore, catalog number: SLHV033RS )
- 24-well plates (Corning, Falcon®, catalog number: 353047 )
- Slip-tip syringe (BD, catalog number: 302833 )
- Sterile cell scraper (Corning, Falcon®, catalog number: 353085 )
- Adult mice (Mus musculus; 6-8-weeks old) (see Notes 7 and 8)
- 2,2,2-tribromoethanol (Avertin) (Sigma-Aldrich, catalog number: T48402 )
- 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) (Thermo Fisher Scientific, GibcoTM, catalog number: 25200056 )
- Phosphate buffered saline (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023 )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153 )
- Primary antibody anti-Pax7 (mouse) (Developmental Studies Hybridoma Bank, catalog number: Pax7 )
- Primary antibody anti-MyoD (rabbit) (Santa Cruz Biotechnology, catalog number: sc-304 )
- Primary antibody anti-MyHC (mouse) (Developmental Studies Hybridoma Bank, catalog number: MF-20 )
- Secondary antibody goat anti-rabbit Alexa Fluor® 488 conjugate (Thermo Fisher Scientific, Invitrogen, catalog number: A-11034 )
- Secondary antibody goat anti-mouse Alexa Fluor® 568 conjugate (Thermo Fisher Scientific, Invitrogen, catalog number: A-11004 )
- 100% ethanol (Decon Labs, catalog number: 2701 )
- Matrigel (Corning, catalog number: 354234 )
- Dulbecco’s modified Eagle’s medium (DMEM) high glucose, pyruvate (Thermo Fisher Scientific, GibcoTM, catalog number: 11995065 )
- Collagenase II (Worthington Biochemical, catalog number: LS004176 )
- Ultra-pureTM water (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977015 )
- Penicillin-streptomycin (Pen/Strep) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) (1 M) (Thermo Fisher Scientific, GibcoTM, catalog number: 15630080 )
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10437028 )
- Recombinant human fibroblast growth factor-basic (bFGF) (PeproTech, catalog number: 100-18B )
- Tris base (Fisher Scientific, catalog number: BP152-5 )
- F-10 Nutrient mixture (Thermo Fisher Scientific, GibcoTM, catalog number: 11550043 )
- Dulbecco’s modified Eagle’s medium (DMEM) (ATCC, catalog number: 30-2002 )
- Horse serum (Thermo Fisher Scientific, GibcoTM, catalog number: 26050088 )
- Dimethyl sulfoxide (DMSO) (Fisher Scientific, catalog number: BP231-100 )
- Paraformaldehyde (PFA) (Sigma-Aldrich, catalog number: P6148 )
- 100% Triton X-100 (Fisher Scientific, catalog number: BP151-500 )
- 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich, catalog number: D8417 )
- 70% ethanol (see Recipes)
- 10% Matrigel (see Recipes)
- Collagenase II (see Recipes)
- Digestion medium (see Recipes)
- Collection/washing/mincing solution (see Recipes)
- Neutralization/isolation media (see Recipes)
- Basic fibroblast growth factor (bFGF) (see Recipes)
- Myoblast growth medium (MGM) (see Recipes)
- Post isolation washing medium (see Recipes)
- Differentiation medium (DM) (see Recipes)
- Freezing medium (see Recipes)
- 4% paraformaldehyde (PFA) (see Recipes)
- 0.3% Triton X-100 (see Recipes)
- 10% Triton X-100 (see Recipes)
- Blocking solution (see Recipes)
- DAPI (see Recipes)
Equipment
- Autoclave
- Dissection tools: Sterilized/autoclaved scissors and forceps
- Biosafety cabinet (Thermo Fisher Scientific, Thermo ScientificTM, model: 1300 Series Class II , Type A2)
- Bench top centrifuge for 1.5 ml Eppendorf tubes (Eppendorf, model: 5424/5424 R )
- Bench top centrifuge for 15 ml and 50 ml tubes (Eppendorf, model: 5702/5702 R/5702 RH )
- Heated incubator shaker (Eppendorf, New BrunswickTM, model: Excella E24 )
- CO2 incubator (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3578 )
- Microscope (Nikon Instruments, model: Eclipse TE2000 )
- Water bath (Thermo Fisher Scientific, model: Model 215 , catalog number: 15-462-15Q)
- 500 ml bottle
Procedure
The basic steps for isolation and purification of primary myoblasts from hind limb muscle of mice are presented in Figure 1.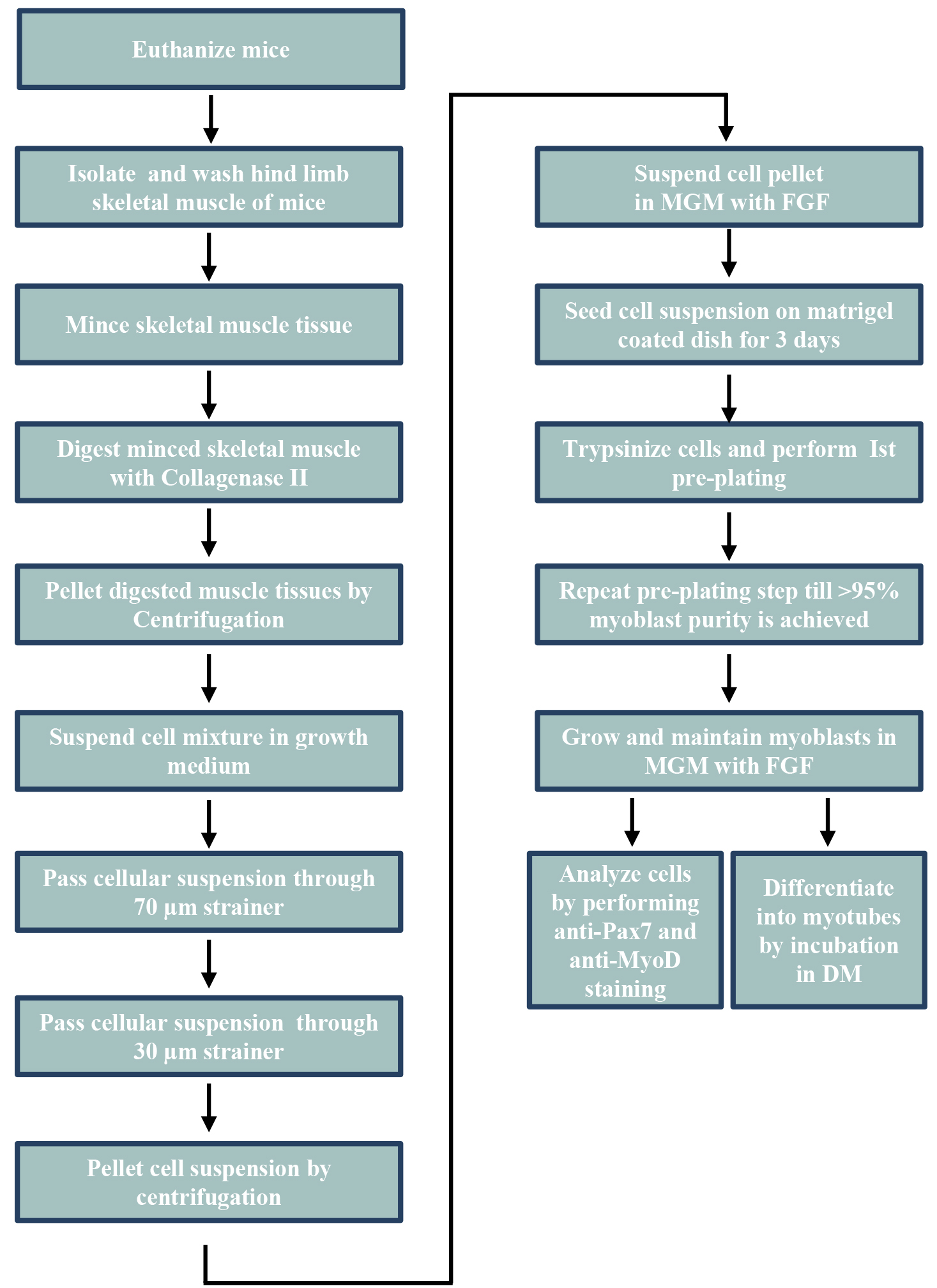
Figure 1. The schematic view of general procedures for the isolation of primary myoblasts from hind limb muscle of mice. MGM, myoblast growth medium; FGF, fibroblast growth factor; DM, differentiation medium.
- Before isolation of primary myoblasts
- Autoclave two sets of tools in sterilization pouches each containing one small pair of scissors and forceps (see Notes 1 and 2).
- Following Recipe 1, prepare 70% ethanol.
- Following Recipe 2, prepare 10% Matrigel (see Note 3).
- Following Recipe 3, prepare collagenase II (see Note 4).
- Following Recipe 4, prepare base of digestion medium (see Notes 5 and 6).
- Following Recipe 5, prepare collection/washing/mincing solution.
- Following Recipe 6, prepare neutralization/isolation medium (see Note 5).
- Following Recipe 8, prepare myoblast growth medium (MGM) (see Note 5).
- Coat 100 x 20 mm Petri dishes (one plate per mouse) by pouring and spreading 10% Matrigel solution enough to cover the plate area. Avoid bubbles. After 30 sec, remove excess Matrigel solution (excess 10% Matrigel can be collected and stored for re-use multiple times, if kept sterile). Leave the plate without cap to dry in sterile air flow in a biosafety cabinet (~20 min). Once dried, put lid on the plate and leave in sterile biosafety cabinet at room temperature until ready to use. There is no need to turn on UV light of biosafety cabinet. This plate will be used for isolation and culturing of myoblasts following digestion of muscle with collagenase.
- For muscle collection and washing before digestion, prepare a 6-well plate containing 3 ml/well of collection/washing/mincing solution (only 4 wells of a 6-well plate needed per mouse).
- For muscle mincing following isolation, prepare two 1.5 ml Eppendorf tubes per mouse each containing 500 µl of collection/washing/mincing solution.
- Autoclave two sets of tools in sterilization pouches each containing one small pair of scissors and forceps (see Notes 1 and 2).
- Muscle removal and washing
- Euthanize mouse following approved IACUC guidelines in your laboratory. For purpose of this protocol, the mouse was given intraperitoneal injection of 2,2,2-tribromoethanol at a dose of 250 mg/kg, followed by cervical dislocation.
- The isolation of hind limb muscles from mice is performed outside a biosafety cabinet. For this step, no sterile techniques are necessary except autoclaved dissection tools and spray of ethanol. Spray hind-limbs with 70% ethanol and pin the mouse face up.
- Remove skin using autoclaved scissors from both hind-limbs to expose muscles.
- Using the first pair of autoclaved scissors and forceps, collect tibial anterior, gastrocnemius, soleus, quadriceps and extensor digitorum longus muscles from one hind-limb and place them into one well of 6-well plate containing collection/washing/mincing solution (Video 1).
 Video 1. Isolation of hind limb muscles from mouse. This video shows basic steps for isolation of hind limb muscles from euthanized mouse and transferring them into a 6-well plate containing collection/washing/mincing solution.
Video 1. Isolation of hind limb muscles from mouse. This video shows basic steps for isolation of hind limb muscles from euthanized mouse and transferring them into a 6-well plate containing collection/washing/mincing solution. - Repeat the previous step on the other hind limb and place the muscles in the second well of 6-well plate containing collection/washing/mincing solution (see Note 9).
- Transfer the muscles using the second pair of autoclaved forceps to the other two wells of the 6-well plate filled with collection/washing/mincing solution. This is needed to wash and remove any sticking hair or unwanted tissue on isolated muscles.
- Put the lid on the 6-well plate containing muscle and collection/washing/mincing solution and spray with 70% ethanol before transferring the plate to a sterile biosafety cabinet. From this step onwards, all the procedure must be performed in biosafety cabinet to avoid potential contamination of myofibers.
- Euthanize mouse following approved IACUC guidelines in your laboratory. For purpose of this protocol, the mouse was given intraperitoneal injection of 2,2,2-tribromoethanol at a dose of 250 mg/kg, followed by cervical dislocation.
- Muscle mincing/digestion
- Transfer washed muscles using second pair of autoclaved forceps to two 1.5 ml Eppendorf tubes (one tube for each hind limb muscle group) containing collection/washing/mincing solution and mince muscles very finely using second pair of autoclaved scissors (see Note 10).
- Once muscles are minced, spin-down tubes using an RT centrifuge at 21,130 x g for 30 sec to separate the minced muscle pieces from the collection/washing/mincing solution.
- Set spun tubes aside under sterile biosafety cabinet.
- Complete preparation of digestion media by adding 1 ml of collagenase II to base of digestion media prepared previously (see Note 6). Filter the entire digestion mixture using a 0.22 μm syringe filter into a 15 ml tube.
- Remove the supernatant from the Eppendorf tubes containing the minced muscles (in step C2).
- To transfer the minced muscles to digestion media tube, cut off the tip of a sterile 1 ml pipette tip to create a large bore to accommodate minced muscles’ size.
- Transfer a small amount of digestion media to the Eppendorf tubes to resuspend minced muscle and then transfer the mix (minced muscles + digestion media) to the original 15 ml digestion media tube (in step C4).
- Repeat step C7 until all the minced muscles are transferred from the Eppendorf tubes to the 15 ml tube containing digestion mixture.
- Close the lid of the 15 ml tube containing minced muscle and digestion media and wrap the lid with Parafilm to avoid any potential leakage.
- Vortex tube for 5 sec.
- Place tube horizontally in a 37 °C shaker and shake at 100 rpm for 1 h with another 5 sec vortex half way through (see Note 11).
- After 1 h, mix should appear cloudy and muscle pieces should be mostly digested and very small (see Note 12).
- Once digestion is complete, vortex tube containing digested muscle mixture for 5 sec.
- Spin the tubes at 1,400 x g for 5 min at room temperature.
- Under sterile biosafety cabinet, remove supernatant (digestion media) and discard.
- Resuspend digested muscle pellet in 7 ml of neutralizing/isolation media using a 10 ml serological pipette. Pipet the digested muscle tissue pellet up and down for 20-30 times using a sterile 10 ml pipette. This ensures that most myoblasts are released from the muscle tissues.
- Place a 70 μm strainer on a 50 ml tube.
- Pre-wet the 70 μm strainer with 2 ml of neutralizing/isolation media.
- Collect resuspended muscle pellet mixture (step C16) using a 10 ml serological pipette and pass through the pre-wet 70 μm strainer.
- Wash the strainer with 2 ml of neutralizing/isolation media to ensure that cells are sticking to strainer (most tissue debris will be removed during this step).
- In a 15 ml tube, pre-wet a 30 μm filter with 1 ml of neutralizing/isolation media.
- Pass cell mixture (previously strained through 70 μm strainer) through the pre-wet 30 μm filter. This step will remove large infiltrating cells such as macrophages from the myoblasts.
- Wash filter with 1 ml of neutralizing/isolation media to ensure that cells are not sticking to the strainer.
- Spin tubes containing cellular mixture at 1,400 x g for 5 min.
- Remove supernatant (neutralizing/isolation media) and resuspend cell pellet in 10 ml MGM + bFGF.
- Seed the cellular mixtures onto the 10% Matrigel pre-coated dish.
- Visualize the cells under a microscope. At this step, there will be a heterogeneous mixture of muscle particles and cells (Figure 2).
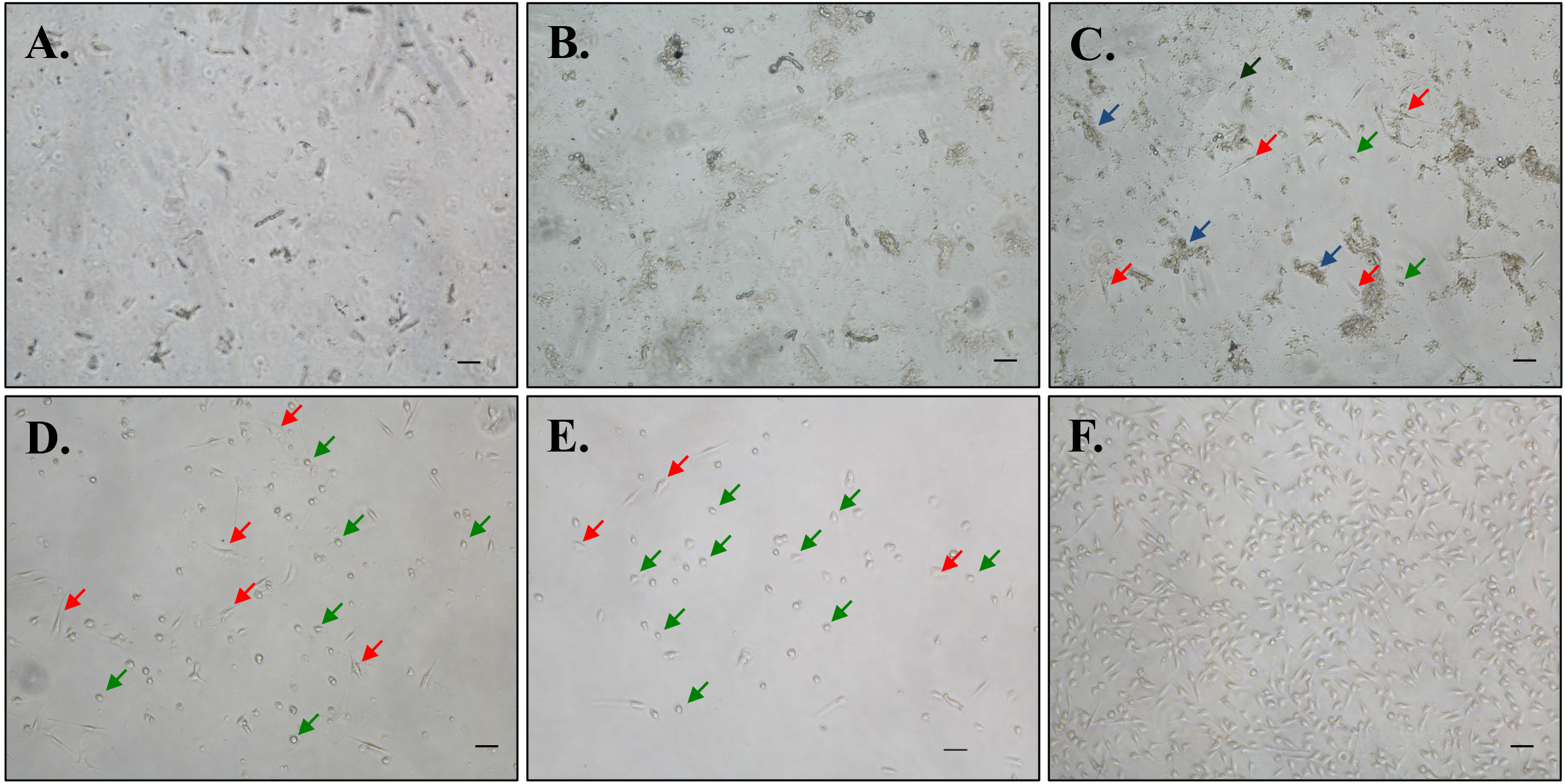
Figure 2. Representative phase contrast microscopy images of primary myoblast cultures at various stages of purification. Digested muscle mixture following filtration at seeding in 10% Matrigel-coated culturing dish at (A) 0 h, (B) 24 h, and (C) 72 h. Appearance of myoblasts after (D) first pre-plating and (E) second pre-plating steps. F. Purified myoblast cultures after 2-4 pre-plating steps. Scale bars = 20 µm. Blue arrows point to tissue/cellular debris; Red arrows point to fibroblasts (large and flat triangular-shaped) and green arrows point to myoblasts (round or bipolar spindle-shaped). - Keep the dish in a 37 °C CO2 incubator for 72 h. Do not change medium during this incubation period.
- After 72 h, you should be able to visualize adherent, small, droplet-shaped myoblasts and large triangular-shaped fibroblasts in addition to remnant tissue debris.
- At this time, the cells are ready for first pre-plating (see Note 13).
- Transfer washed muscles using second pair of autoclaved forceps to two 1.5 ml Eppendorf tubes (one tube for each hind limb muscle group) containing collection/washing/mincing solution and mince muscles very finely using second pair of autoclaved scissors (see Note 10).
- Myoblast purification (Pre-Plating)
- Remove culture media from the dish.
- Using a 10 ml serological pipette, gently wash adherent cells two times with post-isolation media (F-10 medium containing 1% Pen/Strep).
- Detach cells by adding 2 ml of 0.25% trypsin-EDTA and placing cells at 37 °C in a CO2 incubator for 2 min.
- Visualize under the microscope to ensure that all cells are detached.
- Once cells are detached, add 10 ml of MGM + bFGF to dish (see Note 14).
- Transfer cell mixture to a non-Matrigel coated tissue culture dish and place at 37 °C in a CO2 incubator for 45 min. This will allow larger cells such as fibroblasts to adhere in the bottom of the dish while most myoblasts would still be suspended in the media.
- During this time, coat a new Petri dish with 10% Matrigel as described above.
- After 45 min, transfer all supernatant from pre-plating dish to the new Matrigel-coated dish and place the dish in CO2 incubator (see Note 15).
- Pre-plating step should be performed every 36-48 h until > 98% myoblast purity is achieved (see Note 16). Representative phase contrast images at various steps of myoblast isolation are presented in Figures 2C-2E. Purified myoblast culture image is presented in Figure 2F.
- Remove culture media from the dish.
- Culturing of myoblasts
- The purified myoblasts are maintained in culture by incubation in the myoblast growth medium and changing media every other day.
- Do not allow cells to reach > 70% confluency (unless experimentally required) as this will lead to pre-mature differentiation and fusion of myoblasts with each other.
- Expand cells by splitting into multiple 10% Matrigel-coated Petri dishes as needed.
- The myoblasts can be collected in freezing medium and stored in liquid nitrogen tank or -80 °C freezer like any other cultured cells. Primary myoblasts maintain their proliferation and differentiation capacity once thawed and cultured again.
- The purified myoblasts are maintained in culture by incubation in the myoblast growth medium and changing media every other day.
- Differentiating primary myoblasts to myotubes
- Coat tissue culture plates with 10% Matrigel similar to those described above.
- Plate primary myoblasts in pre-coated Matrigel plates. For obtaining good quality myotubes, myoblasts should be at 85-95% confluency before adding differentiation medium (DM). For a 6-well tissue culture plate, we need 0.75-0.9 x 106 cells per well; however, this number will vary depending on the cell passage number. Low passage cells are normally smaller in size therefore use the higher end of the range to achieve desired confluency, whereas higher passage cells are more elongated and larger in size so the lower end of cell number will be required (see Note 17).
- Next day, replace myoblast growth medium with DM (see Note 18).
- DM should be changed every 48 h.
- Myoblasts align and fusion starts within 24-36 h after incubation in DM. At 48 h, small myotubes can be clearly seen, and by 72-96 h they become large and mature.
- Coat tissue culture plates with 10% Matrigel similar to those described above.
- Immunostaining of primary myoblasts and myotubes
- Remove culture media from 6-well tissue culture plates containing primary myoblasts or myotubes which are adhered to surface of the wells.
- Fix adhered cells by adding appropriate amount of 4% PFA to cover cells and incubate 15 min.
- Remove 4% PFA and gently wash the cells three times with 1x PBS for 5 min each.
- Permeabilize the cells by adding 0.3% Triton X-100 and incubate for 7 min at room temperature.
- Remove 0.3% Triton X-100 and gently wash cells three times with PBS for 5 min each.
- Remove PBS and block cells by adding 2% BSA solution for 20 min.
- Prepare primary antibody mixtures in blocking solution.
Note: We use 1:10 dilution of Pax7 antibody, 1:200 dilution of MyoD, and 1:200 dilution of MF-20 (i.e., MyHC) antibody. - Remove blocking solution and add desired primary antibody mixture to each well.
- Wrap plates with Parafilm to prevent evaporation of primary antibody solution.
- Primary antibody incubation can be performed for 1-2 h at room temperature or overnight at 4 °C.
- Once primary antibody incubation period is completed, remove primary antibody solution and wash cells three times with PBS for 5 min each.
- Prepare secondary antibody mixture in blocking solution at a concentration of 1:1,500 for each secondary antibody.
Note: We use anti-mouse Alexa 568 for red fluorescence and anti-rabbit Alexa 488 for green fluorescence. - Wrap plates with Parafilm to prevent evaporation of secondary antibody.
- Secondary antibody incubation is performed at room temperature for 1 h in the dark.
- Remove secondary antibody solution and wash cells three times with PBS for 5 min each.
- Prepare DAPI working solution in PBS.
- Remove PBS from the wells and add DAPI solution for staining nuclei.
- DAPI incubation is performed at room temperature for 3 min in the dark.
- Remove DAPI and wash the cells three times with PBS for 5 min each.
- Add PBS to the wells.
- The cells are now ready to be visualized under a fluorescent microscope. Representative images of myoblast culture after staining with anti-Pax7, anti-MyoD, and DAPI are presented in Figure 3. Differentiated myotube culture images after staining with anti-MyHC and DAPI are presented in Figure 4.
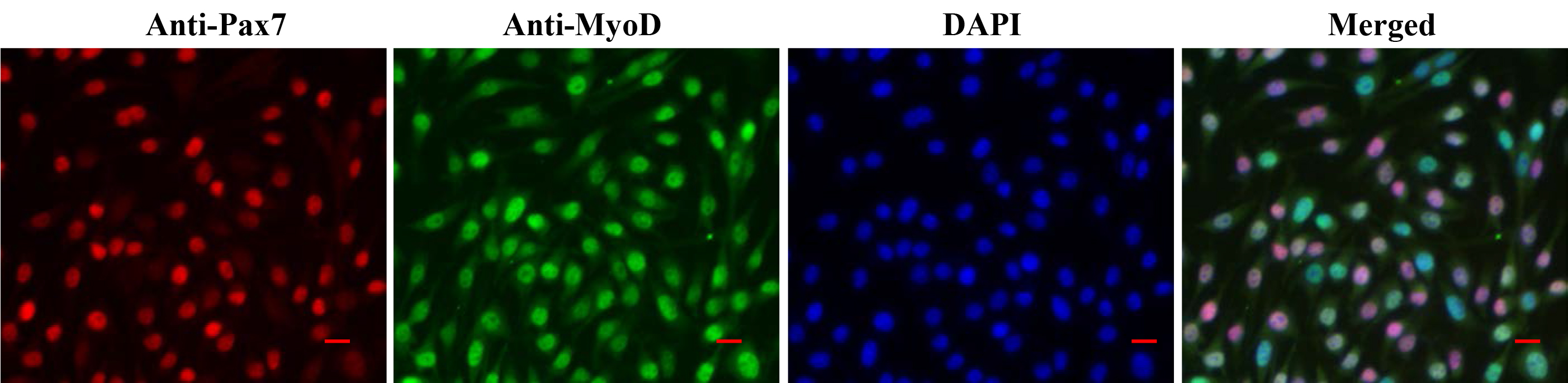
Figure 3. Staining of primary myoblast culture for Pax7 and MyoD. Primary myoblast cultures grown in MGM were fixed and immunostained for Pax7 and MyoD protein. Nuclei were counterstained with DAPI. Representative individual anti-Pax7, anti-MyoD, and DAPI-stained and merged images (triple stained) are presented here. Scale bars = 20 µm.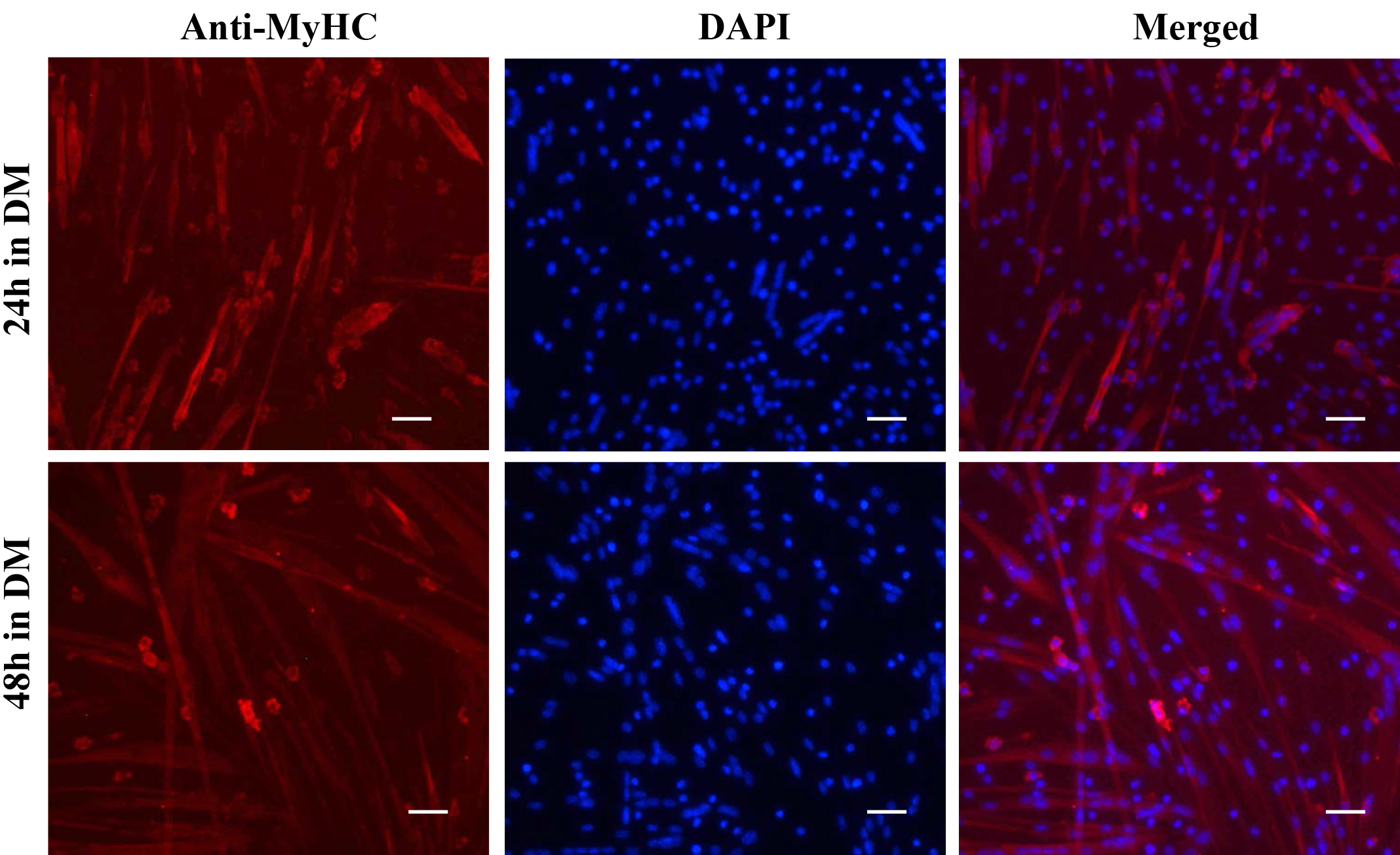
Figure 4. Staining of primary myotube cultures for myosin heavy chain (MyHC). Primary myoblasts were incubated in DM for 24 h or 48 h. The cultures were fixed and immunostained for MyHC protein. Nuclei were counterstained with DAPI. Representative individual anti-MyHC and DAPI-stained and merged images are presented here. Scale bars = 20 µm.
- Remove culture media from 6-well tissue culture plates containing primary myoblasts or myotubes which are adhered to surface of the wells.
Data analysis
For individual experiments, the sample size should be determined by power analysis. For most of our studies focused on studying the effect of regulatory factors on myoblast proliferation and differentiation, we perform experiments in 4-5 replicates. For studying myogenic differentiation, we calculate myogenic index which is percentage of nuclei in MyHC-stained myotubes in total nuclei in the plate. We present the data as mean ± standard deviation (SD). We use paired or unpaired Student’s t-test to determine statistical differences among different groups similar to as described (Ogura et al., 2015; Hindi and Kumar, 2016). A P < 0.05 is considered as statistically significant.
Notes
- All steps except initial isolation of hind limb muscle should be performed under sterile conditions. Even for the isolation of hind limb muscle from euthanized mice, use of autoclaved dissection tools and spray ethanol is highly recommended.
- One set will be used to collect muscle from mouse while the other set will be used under the sterile biosafety cabinet to transfer and mince muscle.
- Do not allow 100% Matrigel to reach RT as this will cause it to solidify and form a thick gel like material that would be difficult to dilute in DMEM.
- Worthington’s powder stock of collagenase does not have same activity (i.e., units per mg dry weight) in all the batches. Therefore, carefully read the activity present in the stock powder vial when preparing collagenase stock each time.
- All media should be warmed at 37 °C before use.
- Collagenase should only be added when muscle is minced and ready to be digested as adding collagenase ahead of time will weaken its digestive activity.
- To obtain primary myoblasts from adult satellite cells mice must be ≥ 6 weeks of age.
- To obtain optimal primary myoblasts mice should not be ≥ 8 weeks of age. This protocol can be performed on younger or older mice if experimental settings require so however some alterations in myogenic yield and purity may be observed; younger mice will yield more myoblasts as their muscle contain actively proliferating cells that are contributing to muscle development and growth. On other hand, older or diseased mice may yield less myoblasts with lower purity and higher fibroblast content. Similar consideration should be taken into account when isolating primary myoblasts from genetic mouse models or mice subjected to various experimental procedures.
- Muscle collection from both limbs should be performed within 5 min to preserve viability.
- Make sure that muscles are finely minced at this step as this will affect the yield of myoblasts.
- It is important to observe the appearance of the digesting muscle mixture at this point. Muscle pieces should look smaller and the digestion medium should start to appear cloudy. If muscle pieces are very small then reduce the second half of the digestion time to 20 min.
- If muscle pieces still appear larger in size and do not seem to be completely digested, extend the digestion time for another 10 min or until digestion appears to be complete.
- If cell density is very low 72 h post isolation, do not pre-plate at this time, just wash dish with F-10 + 1% PS and re-feed with fresh MGM + bFGF.
- bFGF is either added directly to the culturing dish at a concentration of 10 ng/ml, or a mixture of MGM + bFGF can be prepared ahead of time at a similar concentration, however this is only good for 1-week if stored properly at 4 °C.
- Sometimes myoblasts also settle to the bottom of the pre-plating dish, for that reason add media to that dish as well then next day if a large amount of myoblasts is observed repeat the pre-plating step for a shorter duration (~20 min).
- Although high myoblast purity can usually be achieved by performing 1-4 pre-plating repetitions, under certain circumstances this does not suffice. If myoblasts do not seem to be increasingly dominating the culture with each pre-plating step then one can resolve to an alternative purification strategy:
- Split your cultures into multiple Petri dishes so that each dish contains a cellular mixture that is around 10% confluent.
- Each cell should appear spatially separated from its neighboring cells.
- To allow cells to grow continuously change MGM every other day.
- With each day you should observe the formation of myoblast colonies originating from a single cell. You will also observe the formation of fibroblast colonies originating from a single cell.
- Under an inverted microscope, locate the areas where fibroblast colonies are observed and label those areas by marking the bottom of the dish using a marker.
- Under the sterile biosafety cabinet, physically scrape off the fibroblast colonies (marked in Note 16e) using a sterile cell scraper.
- Aspirate the culturing medium and wash your dish a couple of times with post-isolation washing medium.
- Trypsinize the remaining cells (should be pre-dominantly myoblast colonies) left over after scraping and washing off fibroblast colonies in Notes 16f and 16g.
- Plate your cells in a newly coated 10% Matrigel dish.
- High myoblast purity should be observed.
- Avoid overcrowding the well with cells when plating for differentiation, as this will restrict the space that is required for myoblasts to elongate prior to fusion and impose extra stress on the cells, possibly leading to cell death and defective myotube formation.
- If cells are not at optimum confluency next day, allow cells to proliferate longer to achieve desired confluency before switching to DM.
Recipes
- 70% ethanol
Combine 700 ml of 100% ethanol with 300 ml of deionized water - 10% Matrigel
- Thaw Matrigel in ice overnight
- Dissolve 10 ml cold Matrigel in 90 ml of cold DMEM
- Aliquot 5 ml of 10% Matrigel per 15 ml sterile tube into multiple tubes for later use
- Store 10% Matrigel aliquots at -20 °C for long term storage
- For frequent use, store 10% Matrigel tube at 4 °C
- Thaw Matrigel in ice overnight
- Collagenase II
- Prepare stock by dissolving collagenase II in ultra-pure water at a concentration of 4,000 U/ml
- Distribute this collagenase II stock solution into multiple 1 ml aliquot in sterile Eppendorf tubes for later use
- Store aliquots at -20 °C
- Thaw collagenase II immediately before use
- Prepare stock by dissolving collagenase II in ultra-pure water at a concentration of 4,000 U/ml
- Digestion medium
10 ml digestion media is enough for digesting hind limb muscle of one mouse, volume can be modified if older mice are used
For 10 ml: - Prepare base of digestion medium, by supplementing 8.65 ml DMEM (Gibco) with 100 μl of Pen/Strep (1%) and 250 μl of HEPES (2.5%). Keep at 37 °C in a water bath until ready for use
- Once muscle has been isolated, minced and ready for digestion, add collagenase II to make a final concentration of 400 U/ml. Shake briefly by hand
- Filter digestion medium using a 0.22 μm filter
- Collection/washing/mincing solution
For 15 ml, combine 150 μl of Pen/Strep with 14.85 ml of 1x PBS to achieve a solution of 1% Pen/Strep in PBS - Neutralizing/isolation medium
To prepare a 500 ml stock of neutralizing medium: - Remove 50 ml of DMEM (Gibco) from 500 ml bottle and store at 4 °C for any later uses
- Add 50 ml of filtered FBS to DMEM bottle now containing 450 ml DMEM to achieve a concentration of 10% FBS
- Add 5 ml of Pen/Strep to above mix to achieve a concentration of 1% Pen/Strep
- Store this stock at 4 °C
- Warm desired amount at 37 °C before use
- Basic fibroblast growth factor (bFGF)
- Dilute 50 μg of bFGF (50 μg/ml) in 1 ml of filter-sterilized 5 mM Tris (pH 7.6) with 0.1% BSA
- Aliquot into small (50-100 μl) and store at -20 °C or -80 °C
- Thaw aliquot immediately before use
- Dilute 50 μg of bFGF (50 μg/ml) in 1 ml of filter-sterilized 5 mM Tris (pH 7.6) with 0.1% BSA
- Myoblast growth medium (MGM)
To prepare a 500 ml stock of MGM: - Remove 100 ml of F-10 media from a 500 ml bottle and store the removed media at 4 °C for any potential use
- Add 100 ml of filtered FBS to 400 ml F-10 medium. Add 5 ml of Pen/Strep solution
- Store the media at 4 °C
- Warm desired amount of this media at 37 °C before use
- Add 10 ng/ml basic fibroblast growth factor (bFGF) just before using the media for cells (Note 12)
- Each dish will require 10 ml of MGM
- Post-isolation washing medium
For 50 ml:
Supplement 49.5 ml of F-10 media with 500 μl of Pen/Strep to achieve a concentration of 1% Pen/Strep
This can be stored at 4 °C. Warm at 37 °C before use - Differentiation medium (DM)
For 500 ml stock: - Remove 15 ml of DMEM (ATCC) from a 500 ml bottle
- Add 10 ml of filtered horse serum to the DMEM bottle
- Add 5 ml of Pen/Strep to above mixture to achieve a concentration of 1% Pen/Strep
- Store this stock at 4 °C
- Warm desired amount at 37 °C before use
- Freezing medium
For 10 ml:
Combine 5 ml of MGM (see Recipe 8) with 4 ml of filtered FBS (40% FBS) and 1 ml of DMSO (10%)
This can be stored at 4 °C and warmed just before use - 4% paraformaldehyde (PFA)
For 10 ml: - Suspend 0.4 g of PFA in 10 ml of PBS
- Shake at 250 rpm at 60 °C for 1-2 h until completely dissolved
- Allow to cool at room temperature and filter by passing through a 0.45 μm syringe filter
- Store at 4 °C
- For optimal results, do not use 4% PFA if stored more than 2 days
- 0.3% Triton X-100
- Prepare a stock of 10% Triton X-100 by combining 1 ml of 100% Triton X-100 with 9 ml of PBS
- To prepare 10 ml of 0.3% Triton X-100, add 300 μl of 10% Triton X-100 to 9.7 ml PBS
- Blocking solution
For 50 ml: - Dissolve 1 g BSA in 50 ml of PBS
- Filter using a 0.45 μm syringe filter
- This can be stored at -20 °C for future use
- DAPI
For 10 ml:
Add 2 μl of DAPI stock solution to 10 ml of PBS and filter using a 0.45 μm syringe filter
The working solution of DAPI can be stored in the dark at 4 °C
Acknowledgments
This work was supported by funding from NIH grants AR059810, AR068313, and AG029623 (to A. Kumar) and AR069985 (to S. M. Hindi). This protocol has been adapted and slightly modified from previously published articles (Rando and Blau, 1994; Hindi et al., 2014; Ogura et al., 2015; Hindi and Kumar, 2016).
References
- Bentzinger, C. F., Wang, Y. X. and Rudnicki, M. A. (2012). Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4(2).
- Bohnert, K. R., Gallot, Y. S., Sato, S., Xiong, G., Hindi, S. M. and Kumar, A. (2016). Inhibition of ER stress and unfolding protein response pathways causes skeletal muscle wasting during cancer cachexia. FASEB J 30(9): 3053-3068.
- Buckingham, M., Bajard, L., Chang, T., Daubas, P., Hadchouel, J., Meilhac, S., Montarras, D., Rocancourt, D. and Relaix, F. (2003). The formation of skeletal muscle: from somite to limb. J Anat 202(1): 59-68.
- Dogra, C., Changotra, H., Mohan, S. and Kumar, A. (2006). Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-kappaB and degradation of MyoD protein. J Biol Chem 281(15): 10327-10336.
- Hindi, S. M. and Kumar, A. (2016). TRAF6 regulates satellite stem cell self-renewal and function during regenerative myogenesis. J Clin Invest 126(1): 151-168.
- Hindi, S. M., Mishra, V., Bhatnagar, S., Tajrishi, M. M., Ogura, Y., Yan, Z., Burkly, L. C., Zheng, T. S. and Kumar, A. (2014). Regulatory circuitry of TWEAK-Fn14 system and PGC-1alpha in skeletal muscle atrophy program. FASEB J 28(3): 1398-1411.
- Ogura, Y., Hindi, S. M., Sato, S., Xiong, G., Akira, S. and Kumar, A. (2015). TAK1 modulates satellite stem cell homeostasis and skeletal muscle repair. Nat Commun 6: 10123.
- Rando, T. A. and Blau, H. M. (1994). Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol 125(6): 1275-1287.
- Simionescu-Bankston, A. and Kumar, A. (2016). Noncoding RNAs in the regulation of skeletal muscle biology in health and disease. J Mol Med (Berl) 94(8): 853-866.
- Yin, H., Price, F. and Rudnicki, M. A. (2013). Satellite cells and the muscle stem cell niche. Physiol Rev 93(1): 23-67.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hindi, L., McMillan, J. D., Afroze, D., Hindi, S. M. and Kumar, A. (2017). Isolation, Culturing, and Differentiation of Primary Myoblasts from Skeletal Muscle of Adult Mice. Bio-protocol 7(9): e2248. DOI: 10.21769/BioProtoc.2248.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Stem Cell > Adult stem cell > Muscle stem cell
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.