- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Robust Generation of Knock-in Cell Lines Using CRISPR-Cas9 and rAAV-assisted Repair Template Delivery
Published: Vol 7, Iss 7, Apr 5, 2017 DOI: 10.21769/BioProtoc.2211 Views: 23220
Reviewed by: Longping Victor TseRajesh B. ThippeshappaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The programmable Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated nuclease 9 (Cas9) technology revolutionized genome editing by providing an efficient way to cut the genome at a desired location (Ledford, 2015). In mammalian cells, DNA lesions trigger the error-prone non-homologous end joining (NHEJ) DNA repair mechanism. However, in presence of a DNA repair template, Homology-Directed Repair (HDR) can occur leading to precise repair of the lesion site. This last process can be exploited to enable precise knock-in changes by introducing the desired genomic alteration on the repair template. In this protocol we describe the delivery of long repair templates (> 200 nucleotides) using recombinant Adeno Associated Virus (rAAV) for CRISPR-Cas9-based knock-in of a C-terminal tag sequence in a human cell line.
Keywords: CRISPR-Cas9Background
Despite numerous reports on knock-out model systems generated by CRISPR-Cas9, knock-in reports are still lagging behind. Because of the many applications, generating knock-in cell lines remains an obvious goal of genome editing. The introduction of knock-in alterations generally relies on the presence of a repair template DNA and activation of the HDR repair mechanism after a site-specific double strand (ds)DNA break is introduced in the genome close to the site of alteration. Different templates can be delivered to the repair machinery ranging from a classical linearized vector containing extensive homology regions and an optional selection cassette, to single strand (ss)DNA oligonucleotides of about 200 nucleotides (Chen et al., 2011). Although ssDNA oligonucleotides are a popular tool, they can only be used to introduce small alterations such as mutations or epitope tags because of DNA synthesis limitations. In addition, the lack of a selection cassette requires robust screening strategies to identify correct clones as no selective pressure is applied on the HDR process. Successful use of integration-deficient rAAV for homologous recombination was already shown before the availability of tailored nucleases (Khan et al., 2011). Both its efficient delivery and ssDNA genome make rAAV a powerful tool to deliver donor repair templates for homologous recombination. Moreover, the secondary structures at the end of the ssDNA molecule block exonuclease activity and stabilize the donor DNA. Even without the use of specific nucleases, knock-in efficiencies of up to 0.7% were obtained in fibroblasts cells (Russell and Hirata, 1998), which was further increased by introducing selection cassettes.
By combining CRISPR-Cas9 with rAAV-mediated repair template delivery, knock-in cell lines can be generated in a robust manner with efficiencies well beyond 50% when selection cassettes are used. This protocol describes the complete procedure for epitope tagging of a gene of choice in the HCT116 colon carcinoma cell line using CRISPR-Cas9 and rAAV. A timeline for the complete experimental procedure is shown in Figure 1.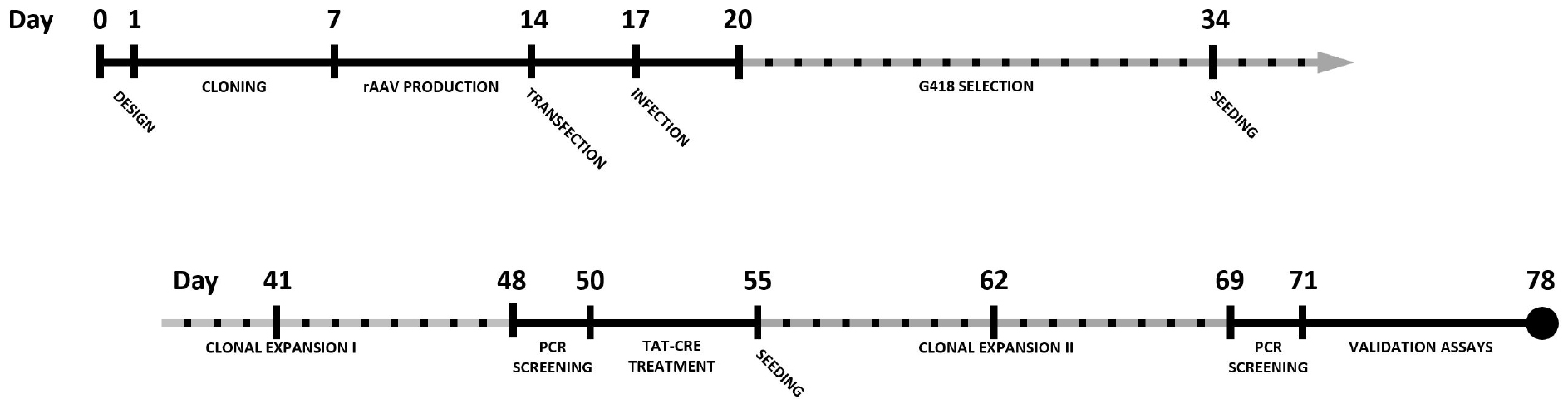
Figure 1. Timeline for the generation of a knock-in cell line using CRISPR-Cas9 and rAAV-assisted repair template delivery. Dotted timespans indicate periods of incubation or expansion, requiring limited to no hands-on time.
Materials and Reagents
- T25 cell culture treated flasks with filter caps (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 136196 )
- Eppendorf Safe-Lock microcentrifuge tubes (Eppendorf, catalog number: 022363204 )
- 5Prime Phase Lock gel tubes heavy 2 ml (Quantabio, catalog number: 2302830 )
- T75 cell culture treated flasks with filter caps (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 178905 )
- Greiner 50 ml centrifuge tubes (Greiner Bio One International, catalog number: 227261 )
- Greiner cell scrapers (Greiner Bio One International, catalog number: 541081 )
- 24-well cell culture-treated multidish (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 142475 )
- 96-well microplates (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 156545 )
- 96-well PCR plate (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: AB0700 )
- 6-well cell culture-treated multidish (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 140675 )
- Aluminium foil tape (3M, catalog number: 425DWB )
- Illustra Microspin S-400 HR columns (GE Healthcare, catalog number: 27-5140-01 )
- Immobilon-FL PVDF transfer membrane (EMD Millipore, catalog number: IPFL00010 )
- Whatman 3 MM chr cellulose blotting sheets (GE Healthcare, catalog number: 3030-917 )
- 0.22 µm filter (EMD Millipore, catalog number: SLGV033RS )
- AAV-293 cell line (Agilent Technologies, catalog number: 240073 )
- HCT 116 cell line (ATCC, catalog number: CCL-247 )
- Cas9 D10A nickase mutant (nCas9) (Addgene, catalog numbers: 48140 and 62987 )
- pAav-MCS-PQS1-3xFLAG or pAav-MCS-PQS2-3xHA (Addgene, catalog numbers: 84883 and 84917 respectively)
- pDG rAAV packaging plasmid (PlasmidFactory, catalog number: PF421 )
- Wild-type Cas9 expression vectors (Cas9) (Addgene, catalog numbers: 48138 and 62988 )
- Competent bacterial cells of choice (e.g., TOP10 or DH5α)
- Surveyor Mutation Detection Kit (Integrated DNA Technologies, catalog number: 706025 )
- UltraPure phenol:chloroform:isoamyl alcohol (25:24:1, v/v) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15593031 )
- Sodium chloride (NaCl) molecular biology grade (EMD Millipore, catalog number: 567441 )
- 2-propanol (Sigma-Aldrich, catalog number: 278475 )
- Ethanol (EMD Millipore, catalog number: 100983 )
- AccuPrime Pfx DNA polymerase (Thermo Fisher Scientific, InvitrogenTM, catalog number: 12344-024 )
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 449709 )
- DMEM, high glucose GlutaMAX medium (Thermo Fisher Scientific, GibcoTM, catalog number: 31966047 )
- Phosphate-buffered saline (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023 )
- Benzonase nuclease (Sigma-Aldrich, catalog number: E1014 )
- AAV-purification Vira Kit 3-use (Virapur, catalog number: 003063 )
- AAV helper-free system (Agilent Technologies, catalog number: 240071 )
- McCoy’s 5A medium, modified (Thermo Fisher Scientific, GibcoTM, catalog number: 16600082 )
- Opti-MEM I reduced serum medium, GlutaMAX supplement (Thermo Fisher Scientific, GibcoTM, catalog number: 51985026 )
- Fugene HD transfection reagent (Promega, catalog number: E2311 )
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10500064 )
- Trypsin-EDTA (Thermo Fisher Scientific, GibcoTM, catalog number: 25300096 )
- Puromycin (Sigma-Aldrich, catalog number: P8833 )
- Crystal violet solution (Sigma-Aldrich, catalog number: HT90132 )
- Geneticin/G418 (Thermo Fisher Scientific, GibcoTM, catalog number: 11811031 )
- GoTaq G2 Hot Start polymerase (Promega, catalog number: M7405 )
- dNTP 100 mM PCR grade (Agilent Technologies, catalog number: 200415 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266 )
- Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D2650 )
- TAT-Cre recombinase (Excellgen, catalog number: EG-1001 )
- NucleoSpin Gel and PCR Clean-up Kit (Machery-Nagel, catalog number: 740609 )
- Random Primer DNA Labeling Kit (Takara Bio, catalog number: 6045 )
- [α-32P] dCTP, 50 µCi (PerkinElmer, catalog number: BLU013H250UC )
- Exo-free Klenow enzyme (New England Biolabs, catalog number: M0212S )
- SuRE/Cut buffer H (Roche Diagnostics, catalog number: 11417991001 )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A4503 )
- EcoRI 40 U/µl (Roche Diagnostics, catalog number: 10200310001 )
- Nytran SuPerCharge TurboBlotter Kit (Sigma-Aldrich, catalog number: Z613924 )
Note: This product has been discontinued. - PerfectHyb plus hybridization buffer (Sigma-Aldrich, catalog number: H7033 )
- 4-12% criterion XT Bis-Tris protein gel 18 well, 30 µl (Bio-Rad Laboratories, catalog number: 3450124 )
- Mouse monoclonal anti-FLAG M2 antibody (Sigma-Aldrich, catalog numbers: F3165 )
Or rat monoclonal anti-HA high affinity (Roche Diagnostics, catalog number: 11867423001 ) - Zero Blunt PCR Cloning Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: K270020 )
- NucleoSpin Plasmid EasyPure (Machery-Nagel, catalog number: 740727 )
- Tris ultra-pure grade (MP Biomedicals, catalog number: 02103133 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E5134 )
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 436143 )
- Proteinase K from Tritirachium album > 800 U/ml (Sigma-Aldrich, catalog number: P4850 )
- HEPES (Sigma-Aldrich, catalog number: H4034 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: 255793 )
- Direct lysis reagent (cell) (VIAGEN BIOTECH, catalog number: 301-C )
- CHAPS hydrate (Sigma-Aldrich, catalog number: C5070 )
- cOmplete protease inhibitor (Roche Diagnostics, catalog number: 11697498001 )
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: 30721 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 221465 )
- Sodium citrate tribasic dehydrate [HOC(COONa)(CH2COONa)2·2H2O] (Sigma-Aldrich, catalog number: C8532 )
- SmartLadder (Eurogentec, catalog number: MW-1700-10 )
- Phase Lock lysis buffer (see Recipes)
- TE-buffer (see Recipes)
- 2x HEBS (HEPES-buffered saline) (see Recipes)
- rAAV cell lysis buffer (see Recipes)
- Direct PCR lysis buffer (see Recipes)
- CHAPS lysis buffer (see Recipes)
- Depurination buffer (see Recipes)
- Denaturation buffer (see Recipes)
- Neutralization buffer (see Recipes)
- 20x SSC (see Recipes)
- Wash buffer 1 (see Recipes)
- Wash buffer 2 (see Recipes)
- Wash buffer 3 (see Recipes)
Equipment
- BSL2 cell culture facilities
- Thermoshaker
- Vortex (e.g., IKA, model: MS2 minishaker )
- Inverted microscope for cell culture use (e.g., Carl Zeiss, model: Axiovert 25 )
- Non-circulating water bath
- 250 ml sterile storage bottle (Corning, catalog number: 430281 )
- Thermal cycler instrument (e.g., Bio-Rad Laboratories, model: T100TM Thermal Cycler , catalog number: 1861096)
- Western blotting equipment, e.g.,
Criterion vertical electrophoresis cell (Bio-Rad Laboratories, catalog number: 1656001 )
Criterion blotter with plate electrodes (Bio-Rad Laboratories, catalog number: 1704070 ) - GS gene linker UV chamber (Bio-Rad Laboratories, model: GS Gene LinkerTM UV Chamber )
- Phosphor imager device (e.g., GE Healthcare, model: Typhoon 9200 )
- Required facilities and procedures (e.g., waste disposal procedures) should be in place for using radioactively labeled nucleotides ([α-32P] dCTP)
- Tabletop centrifuge (Eppendorf, model: 5430 R )
Procedure
- Design of guide RNA (gRNA) constructs for the target gene of interest
- Insert the genomic sequence coding for the C-terminal region of the protein of interest in a guide design tool of choice (e.g., crispr.mit.edu). In most cases, an input sequence with a total length of 250 nucleotides surrounding the stop codon harbors enough PAM motifs for the design tool to generate multiple guide sequences.
- Select guides in close proximity of the C-terminus. Bear in mind potential off-target effects when defining the guides. Most design tools provide this information directly by ranking potential guides based on off-target hit-scores in the target genome.
- Clone guides in a Cas9 expression vector of choice. Use of the Cas9 D10A nickase mutant (nCas9, Addgene plasmids #48140 and #62987) is advised to minimize off-target cleavage events. Alternatively, wild-type Cas9 expression vectors are also available on Addgene (plasmids #48138 and #62988). Detailed cloning procedures for the constructs are described by Ran et al. (2013). When using the Cas9 D10A nickase (nCas9) maintain a minimal offset between the guide pairs to ensure optimal efficiency.
Note: It is recommended to assess the cleavage efficiency of multiple gRNAs in a separate experiment. This can be done by transfecting the constructs and performing a mismatch cleavage assay (e.g., Surveyor mismatch cleavage assay) on the selected cell population (Qiu et al., 2004). We usually test 2-3 different gRNAs for each genomic region of interest. In case of using nCas9 it is recommended to test 2-3 pairs of gRNAs.
- Insert the genomic sequence coding for the C-terminal region of the protein of interest in a guide design tool of choice (e.g., crispr.mit.edu). In most cases, an input sequence with a total length of 250 nucleotides surrounding the stop codon harbors enough PAM motifs for the design tool to generate multiple guide sequences.
- Generation of the rAAV targeting construct by PCR amplification of homology regions
A backbone plasmid for C-terminal tagging with a combinatorial tag consisting of the PQS1 peptide (Vandemoortele et al., 2004) and a 3xFlag tag has been deposited in Addgene (plasmid #84883). An alternative version of this targeting construct containing the PQS2 peptide and a 3xHA-tag is also available on Addgene (plasmid #84917). 5’ and 3’ homology regions (HRs) can be inserted in multiple cloning sites present in these plasmids via standard cloning procedures.- Prepare genomic DNA (gDNA) using a phenol extraction and ethanol precipitation protocol
- Detach and pellet 2.5 x 106 cells corresponding to a subconfluent T25 flask in an Eppendorf tube by centrifugation at 500 x g for 5 min at room temperature (RT).
- Add 500 µl gDNA Phase Lock lysis buffer (see Recipes) to the pellet and incubate overnight at 37 °C.
- Prior to adding the lysate, spin down the Phase Lock tubes by short centrifugation. Shake the lysate samples well before transferring to the Phase Lock tube.
- Add 500 µl phenol:chloroform:isoamyl alcohol (25:24:1, v/v) and shake vigorously for 1 min.
- Centrifuge Phase Lock tube for 3 min at 16,000 x g at RT. Transfer aqueous phase into a fresh Eppendorf tube and add 50 µl 3 M NaCl and 500 µl 2-propanol. The genomic DNA can be observed after inverting the tubes several times.
- Centrifuge for 2 min at 16,000 x g at RT. Discard supernatant and add 1 ml 70% ethanol to the DNA pellet. Invert several times and incubate for 5 min at RT. Centrifuge for 2 min at 16,000 x g at RT.
- Discard supernatant. Air dry the pellet at 52 °C for 5 min prior to re-suspending the pellet in 100 µl 10 mM TE-buffer pH 7.5. Allow pellet to dissolve for 1 h by incubating at 65 °C in a thermoshaker. Measure genomic DNA concentration.
- Design primers to amplify the HRs with appropriate overhanging restriction sites for insertion in the backbone plasmid.
Note: It is recommended to use HRs of at least 650 bp. Shorter HRs are reported to substantially reduce homologous recombination potential (Vasileva et al., 2006). Targeting constructs exceeding 4.7 kb in total length between inverted terminal repeat (ITR) sequences will hamper packaging efficiency of rAAV (Wu et al., 2010). - Prepare PCR mix
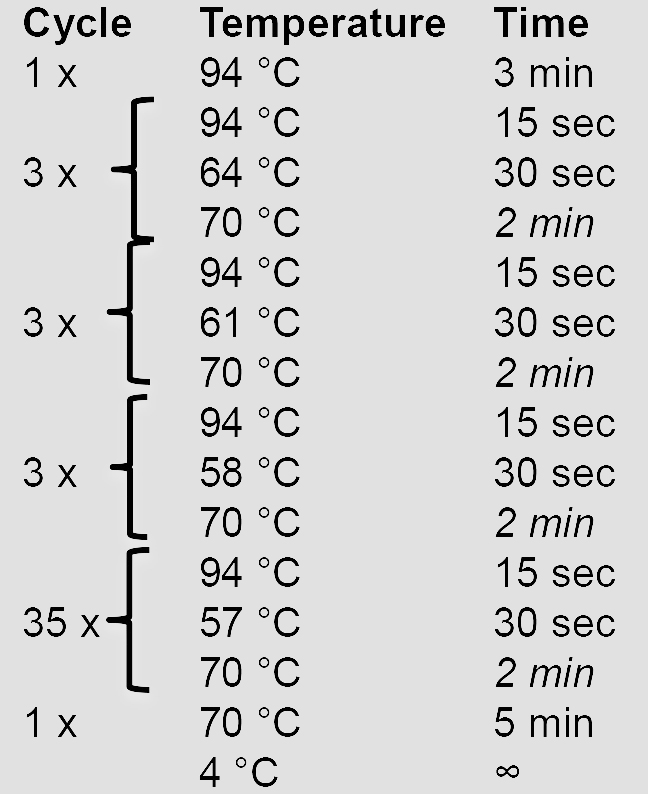
10x AccuPrime Pfx reaction mix 5 µl Forward primer (10 µM) 1.5 µl Reverse primer (10 µM) 1.5 µl AccuPrime Pfx DNA polymerase 0.5 µl gDNA (100 ng/µl) 1 µl ddH2O 40.5 µl - Amplify the HRs using a touchdown PCR program. Elongation times in italics are adjusted according to the length of the HR (1 min/kb).
- Verify and purify the PCR product by agarose gel electrophoresis before proceeding with cloning steps.
Note: As the backbone constructs mentioned above contain multiple cloning sites (MCS) flanking the neomycin selection cassette, cloning of the final targeting constructs can be performed using different restriction enzyme sites. Alternative cloning methods (e.g., Gibson assembly, In-Fusion) are also compatible with these vectors.
- rAAV production and purification
- AAV-293 transfection
- Seed 10 T75 flasks with 4 x 106 AAV-293 cells 24 h before transfection.
- Aspirate and add fresh growth medium at least 30 min prior to transfection.
- Prepare DNA transfection mix for every T75.
pAAV targeting construct (1 µg/µl) 15 µl pDG (1 µg/µl) 30 µl 2.5 M CaCl2 75 µl ddH2O 630 µl
Note: The pDG plasmid serves as a helper plasmid for rAAV packaging. pDG contains all the required AAV and adenoviral functions to ensure amplification and packaging of (r)AAV vectors. - Add DNA transfection mix dropwise to 750 µl 2x HEBS (see Recipes) while using a vortex to ensure thorough mixing. Incubate mixture for 10 min at RT.
- Transfer transfection mix to cells in a dropwise fashion.
Note: Transfected AAV-293 cells and purified rAAV virus should be handled in a BSL-2 facility from this point onwards. - Replace growth medium with 12 ml of fresh DMEM growth medium 24 h after transfection and incubate for an additional 48 h.
Note: Before replacing the growth medium a fine precipitate should be found between the transfected cells by visual inspection using a light microscope. The calcium-phosphate-DNA precipitate aids binding of the DNA to the cell surface before entering the cells by endocytosis. Suboptimal transfection buffers (e.g., 2x HEBS) or DNA qualities can affect precipitate formation leading to low transfection efficiencies. Growth impairment may occur due to transfection.
- Seed 10 T75 flasks with 4 x 106 AAV-293 cells 24 h before transfection.
- Harvest and purification of rAAV particles
- Collect growth medium with detached cells in 50 ml tubes.
Note: Cells may detach during rAAV production. The total amount of growth medium of all T75 Falcons used for production is equally divided over 4 50 ml tubes for convenience in further steps of the purification procedure. - Collect cells in 1.5 ml PBS/T75 flask using a cell scraper and add cell suspensions to the 50 ml tubes. Centrifuge the cells for 3 min at 500 x g and transfer the supernatants to new 50 ml tubes.
- Add 2.5 ml PBS to the obtained cell pellets, re-suspend the cell pellets and combine all cell suspensions in one new tube. Centrifuge for 3 min at 500 x g and re-suspend the cell pellet in 3 ml rAAV lysis buffer.
- Subject pellets to 3 freeze-thaw cycles with brief vortexing between cycles. Transfer the lysate to the supernatant of step C2b.
Note: Unpurified virus can be stored at -80 °C. Tubes should be thawed prior to purification using a 37 °C water bath while swirling the tube to maintain low temperature. - Centrifuge the tubes at 900 x g for 30 min at 4 °C. Collect supernatant in a 250 ml sterile bottle and add Benzonase nuclease (final conc. 50 U/ml).
- Purify rAAV particles using the VIRAPUR AAV Purification Kit according to the manufacturer’s protocol. Purified virus should be stored in aliquots at -80 °C until use.
- Collect growth medium with detached cells in 50 ml tubes.
- AAV-293 transfection
- Transfection and infection of HCT116 cells
- Seed 2.7 x 105 HCT116 cells/well in a 24-well plate containing 1 ml McCoys growth medium.
- Prepare following mixes in separate tubes.
FugeneHD mix DNA mix Opti-MEM 11.7 µl Opti-MEM 11.7 µl Fugene HD transfection reagent 1.4 µl Cas9 + gRNA plasmid DNA 0.5 µg
Gently add the Fugene HD mix to the DNA mix, tap the tube gently and incubate the mixture for 10 min at RT. During this incubation step, replace the medium on the HCT116 cells with 250 µl Opti-MEM.
Note: McCoys growth medium is replaced by Opti-MEM during transfection since cytotoxicity of Fugene-based transfections can increase due to the presence of antibiotics in the growth medium (e.g., penicillin and streptomycin). - Add the Fugene HD-DNA mix dropwise to the HCT116 cells.
- Add 250 µl additional McCoys medium containing 20% FBS to the cells 6 to 8 h after transfection.
- Detach cells with trypsin 24 h post transfection. Transfer half of the cell suspension to a T25 flask. The remainder of the cells can be either discarded or used as non-infected G418 control population described in step D7.
- Dilute 50 µl of rAAV virus in 1 ml McCoys growth medium and add the mixture to the transfected cells 24 h after seeding in T25 flasks.
Note: If colony numbers are markedly lower than expected (Figure 2), infection volumes can be increased to 150 µl or even 500 µl. Selection of cells transfected with CRISPR-Cas9 plasmids by the Zhang lab (see Materials) using puromycin or EGFP-based cell sorting prior to infection is optional. In most experiments, it is sufficient to solely rely on Geneticin/G418 selection for enrichment as this is a direct indicator of rAAV cassette insertion in the target genome. Moreover, treatment of the cells with puromycin or FACS sorting before G418 selection may have a detrimental effect on survival.
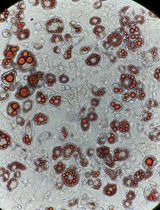
Figure 2. Crystal violet staining of rAAV-infected (left) and non-infected HCT116 cells (right) after 14 days of G418 selection. The crystal violet stain visualizes cell foci resistant to G418. - Add G418 to the cells at a concentration of 1 mg/ml 72 h after transfection.
Note: An additional infected HCT116 population and non-infected HCT116 population is subjected to G418 selection to assess infection efficiency by crystal violet staining of the G418-resistant cell foci or colonies. The expected staining outcome is shown in Figure 2. - Split cells and add fresh G418-supplemented growth medium when culture reaches 70% confluency. After 14 days of selection, seed single HCT116 cells in 96-well plates by manual dilution. Maintain 1 mg/ml G418 selection in the 96-well plates.
Note: In a typical manual dilution step the cells are detached, counted and diluted to 10, 20 and 40 cells/ml. For each density (1, 2 or 4 cells/well) one plate is seeded by pipetting 100 µl of diluted cell suspension in each well of a 96-well tissue culture plate prefilled with 100 µl growth medium. It is recommended to seed different densities to ensure sufficient wells with single cells. - Screen every well for presence of clonal cell populations one week after seeding by visual inspection using light microscopy.
- Once the majority of clonal populations reach approximately 70% confluence (Figure 3) PCR screening of the clonal cell populations can start (Figure 4).
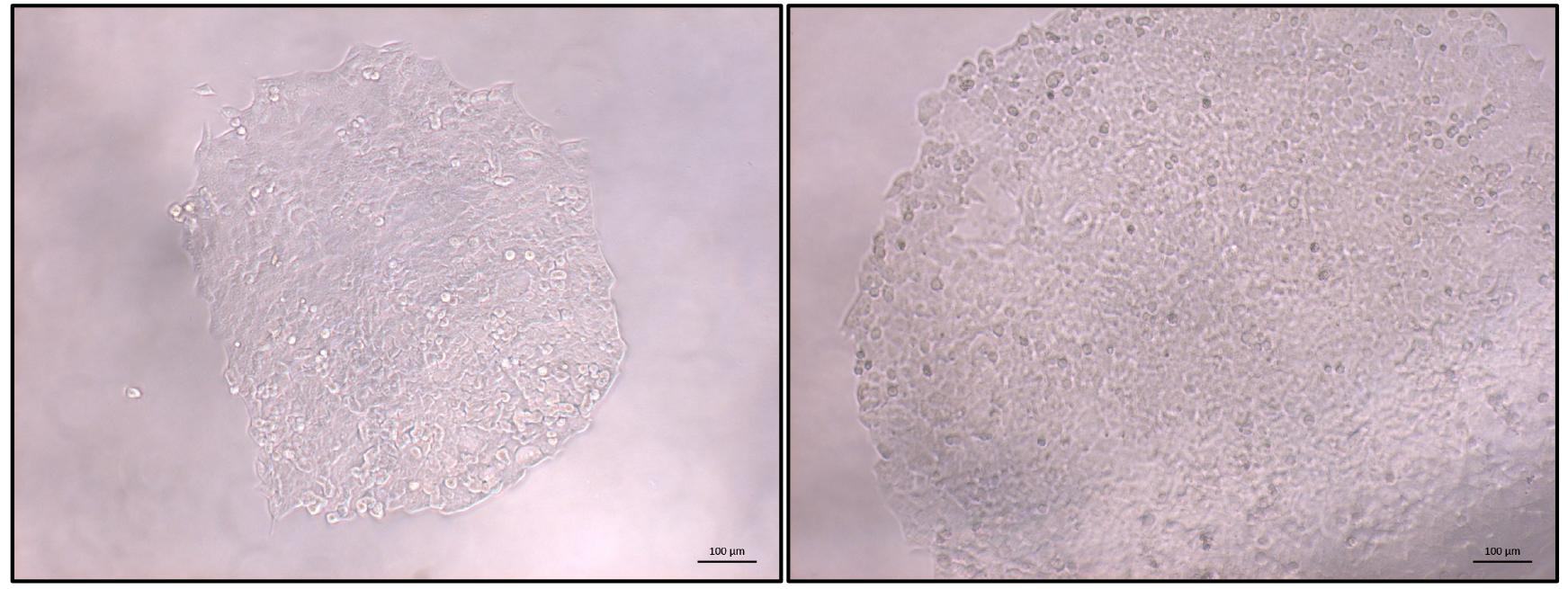
Figure 3. Typical view of an HCT116 clonal population. Once most identified populations reach 70% confluence PCR-based screening can be started. Left: expanding colony; right: colony ready for screening. Scale bars = 100 µm.
- Seed 2.7 x 105 HCT116 cells/well in a 24-well plate containing 1 ml McCoys growth medium.
- PCR-based screening for homology-directed repair events
The different primers used for PCR screening are depicted in Figure 4. It is recommended to design HR screening PCR reactions containing the junction of the HR with the flanking genomic DNA sequence in order to avoid the detection of random integration events (false positives). Random integration events can be detected by Southern blotting (step G2 and Data analysis section).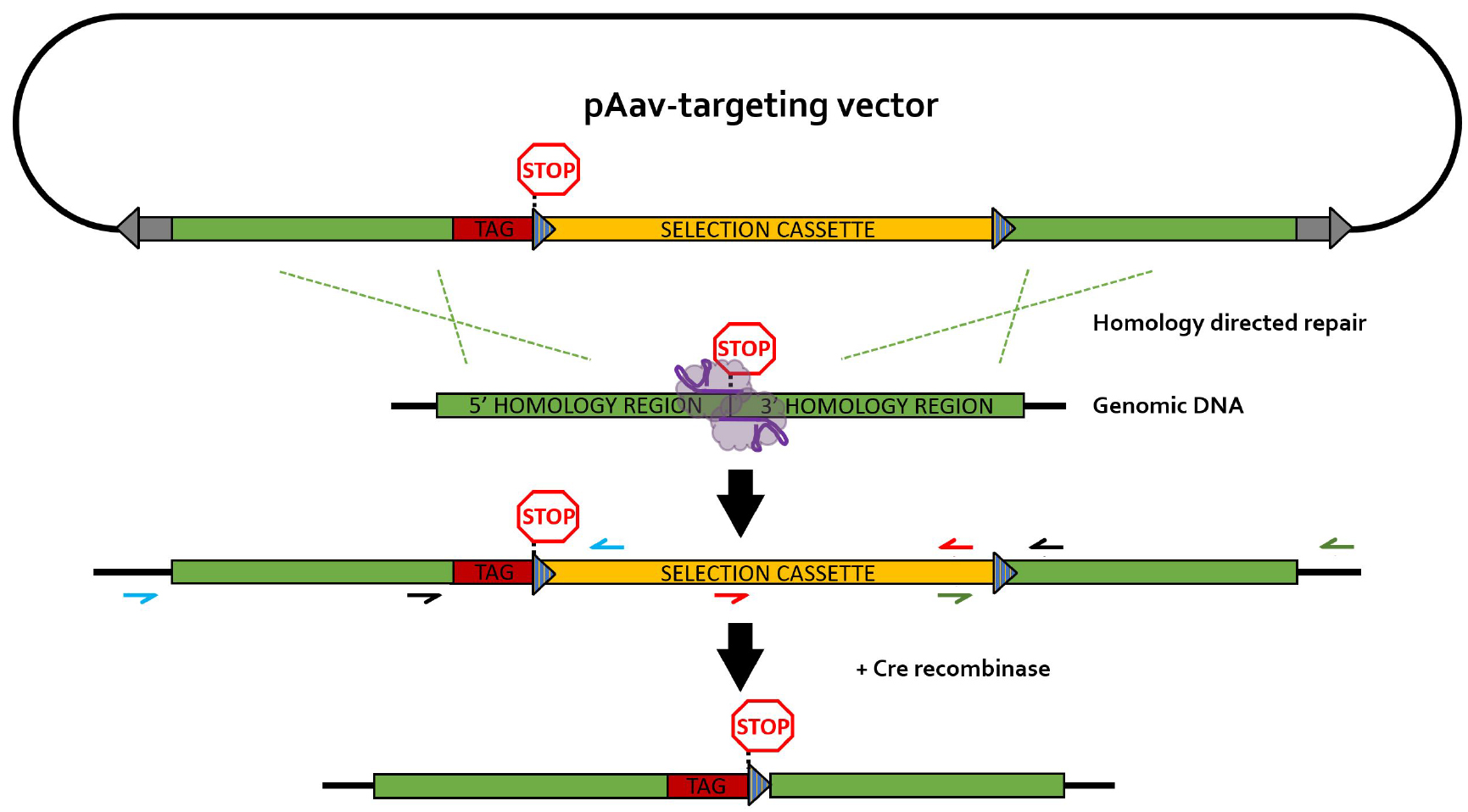
Figure 4. rAAV-mediated homology-directed repair of CRISPR-induced double stranded breaks on a single allele. Different primer pairs used for screening throughout this protocol are indicated in separate colors. Blue primers: 5’HR screening primers; green primers: 3’HR screening primers; black primers: selection cassette removal (CR) screening primer pair; red primers: primer pair for generation of the Southern blot DNA template (SBDT); grey arrows: inverted terminal repeats (ITR); blue striped triangles: loxP sequences; TAG: epitope tag; selection cassette: neomycin-resistance gene under control of the phosphoglycerate kinase (PGK) promotor; purple elements: nickase Cas9 (nCas9) pair.- Generation and lysis of PCR screening plate
- Pipet 20 µl of direct PCR lysis buffer (see Recipes) to a new 96-well PCR plate.
- Remove growth medium from the 96-well plate containing the clonal cell populations. Wash once with PBS before detaching cells in 25 µl trypsin. Transfer 5 µl of the detached cells to the PCR plate prefilled with PCR lysis buffer and cover with aluminium foil tape. Rapidly add 200 µl of standard growth medium to the remaining cell suspension in the 96-well tissue culture plate for further culturing. Place the 96-well PCR plate in a thermal cycler and subject samples to the following program:
15 min 55 °C 45 min 85 °C ∞ 4 °C
Note: After lysis the 96-well PCR plate can be stored at 2-8 °C until PCR screening.
- Pipet 20 µl of direct PCR lysis buffer (see Recipes) to a new 96-well PCR plate.
- PCR amplification.
- Perform a PCR reaction spanning the 5’HR using the lysis material of each clone as input DNA to assess rAAV-mediated integration of the repair template using the program described in step B4.
5x Colorless GoTaq Flexi buffer 4 µl 5’HR Fwd primer (100 µM) 0.15 µl 5’HR Rev primer (100 µM) 0.15 µl dNTP’s (25 mM each) 0.2 µl MgCl2 (25 mM) 1.2 µl DMSO 1.2 µl ddH2O 10.9 µl GoTaq (5 U/µl) 0.2 µl Lysis material 2µl - Assess amplification by agarose electrophoresis. Repeat reaction with 3’HR primers for every positive clone.
5x Colorless GoTaq Flexi buffer 4 µl 3’HR Fwd primer (100 µM) 0.15 µl 3’HR Rev primer (100 µM) 0.15 µl dNTP’s (25 mM each) 0.2 µl MgCl2 (25 mM) 1.2 µl DMSO 1.2 µl ddH2O 10.9 µl GoTaq (5 U/µl) 0.2 µl Lysis material 2 µl - Analyze amplification by agarose electrophoresis.
- Positive clones are expanded and frozen.
- Perform a PCR reaction spanning the 5’HR using the lysis material of each clone as input DNA to assess rAAV-mediated integration of the repair template using the program described in step B4.
- Generation and lysis of PCR screening plate
- Tat-Cre mediated selection cassette removal
- Seed PCR-positive clones from Procedure E in separate wells of a 96-well plate at a density of 1 x 103 cells/well.
- Add TAT-Cre recombinase at a final concentration of 2.5 µM to each well 24 h after seeding.
- Wash twice with DPBS before adding fresh growth medium after 24 h of TAT-Cre incubation. Cells are ready for single cell seeding by manual dilution in 96-well plates 4 days after TAT-Cre treatment.
- Seed PCR-positive clones from Procedure E in separate wells of a 96-well plate at a density of 1 x 103 cells/well.
- Screening and validation of final clones
Note: Assays described in this section are typically performed in a sequential manner.- PCR amplification
- Prepare lysis plate as described in step E1.
Verify excision of the selection cassette by PCR using the program described in step B4. Run this PCR reaction also on untreated positive clones to obtain a reference for clones without successful recombination (see note in step G1c below).5x Colorless GoTaq Flexi buffer 4 µl CR Fwd primer (100 µM) 0.15 µl CR Rev primer (100 µM) 0.15 µl dNTP’s (25 mM each) 0.2 µl MgCl2 (25 mM) 1.2 µl DMSO 1.2 µl ddH2O 10.9 µl GoTaq (5 U/µl) 0.2 µl Lysis material 2 µl - Analyze amplicon size by agarose electrophoresis. In silico removal of the cassette using software for manipulation of DNA sequences can be used to define the expected length of the amplicon.
Note: Successful PCR for the clones without recombination can be difficult to obtain due to size of the amplicon and GC content of the promoter in the neomycin selection cassette. - Southern blot
- PCR amplify a part of the neomycin resistance cassette using the PCR program described in step B4. Purify PCR product with NucleoSpin Gel and PCR Clean-up Kit.
10x AccuPrime Pfx reaction mix 5 µl SBDT forward primer (10 µM) 1.5 µl SBDT reverse primer (10 µM) 1.5 µl AccuPrime Pfx DNA polymerase (2.5 U/μl) 0.5 µl rAAV targeting construct (1 ng/µl) 10 µl ddH2O 31.5 µl SBDT Fwd primer 5’-TGCTCCTGCCGAGAAAGTAT-3’ SBDT Rev primer 5’-GCGATGCAATTTCCTCATTT-3’ - Denature template DNA using Random Primer DNA Labeling Kit.
Template DNA (50 ng/µl) 1 µl Random primer 2 µl ddH2O 11 µl - Boil for 5 min. Snap cool sample on ice for 5 min afterwards. Add following mix to the sample:
10x buffer 2.5 µl dNTP mix (0.2 mM each) 2.5 µl [α-32P] dCTP (50 µCi) 5 µl Exo-free Klenow enzyme 1 µl - Incubate for 20 min at 37 °C. Inactivate the Exo-free Klenow enzyme by incubating at 65 °C for 5 min. Boil sample for 3 min.
Note: Appropriate protective measurements should be taken to work with [α-32P] dCTP and the labeled probe generated using this reagent. - Purify the probe using an Illustra Microspin S-400 HR column according to the protocol provided by the manufacturer.
- Prepare 10 µg 500 ng/µl gDNA of the selected clones as described in step B1. Digest overnight with EcoRI at 37 °C.
gDNA (500 ng/µl) 20 µl SuRE/Cut buffer H 3 µl 0.1% BSA 0.3 µl EcoRI (40 U/µl) 0.5 µl H2O 6.2 µl - Separate digested gDNA by 0.7% agarose gel electrophoresis. Incubate gel in the different buffers (see Recipes) according to following scheme:
Depurination buffer 10 min Denaturation buffer 2 x 15 min Neutralization buffer 2 x 15 min 20x SSC 10 min
Note: Thoroughly rinse both the electrophoresis tank and gel tray with 0.5% SDS and hot water to minimize background signal on Southern blot. Include genomic DNA of clones prior to TAT-Cre treatment for Southern blot as control samples. - Blot overnight using the Nytran SuPerCharge TurboBlotter Kit.
- Wash membrane for 5 min in 2x SSC (see Recipes). Crosslink on Whatman paper (pre-wetted in 2x SSC) in a GS gene linker UV chamber using program C3 (150 mJ by 254 nm UV irradiation).
- Transfer membrane to a tube containing PerfectHyb plus hybridization buffer preheated to 68 °C. Incubate for 1 h at 68 °C.
- Denature radioactive probe by boiling for 10 min. Snap cool on ice for 2 min.
- Add 25 µl of denatured probe to the membrane in fresh preheated PerfectHyb plus buffer. Incubate overnight at 68 °C.
- Rinse membrane three times with wash buffer 1 (see Recipes). Wash membrane according to following scheme.
Wash buffer 1 10 min Wash buffer 2 15 min Wash buffer 3 10 min
Note: Wash buffers 2 and 3 (see Recipes) should be preheated to 65 °C. - Image on phosphorimager device after overnight exposure on a phosphor screen.
- PCR amplify a part of the neomycin resistance cassette using the PCR program described in step B4. Purify PCR product with NucleoSpin Gel and PCR Clean-up Kit.
- Western blot
- Lyse a subconfluent T25 flask of each clone in 100 µl CHAPS lysis buffer (see Recipes). Incubate for 5 min on ice before centrifuging suspension for 10 min > 16,000 x g in a tabletop centrifuge. Transfer supernatant to new tube.
- Measure protein concentration by Bradford assay. Load 50 µg of protein material on SDS-PAGE. After blotting of the proteins on a membrane, modified endogenous proteins can be visualized by (tag-)specific primary antibodies (i.e., anti-FLAG or anti-HA, depending on the chosen backbone plasmid).
- Lyse a subconfluent T25 flask of each clone in 100 µl CHAPS lysis buffer (see Recipes). Incubate for 5 min on ice before centrifuging suspension for 10 min > 16,000 x g in a tabletop centrifuge. Transfer supernatant to new tube.
- Sanger sequencing.
- Prepare gDNA of every clone as described in step B1.
- PCR amplify region of interest using the PCR mix and program described in steps B3 and B4 respectively. Purify PCR fragment and ligate in pCR-Blunt vector using Zero Blunt PCR Cloning Kit.
- Transform in competent bacterial cells of choice (e.g., TOP10 or DH5α) and prepare plasmid DNA using the NucleoSpin Plasmid EasyPure Kit. pCR-Blunt inserts can be sequenced with M13 forward and reverse primers.
M13 forward 5’-TGTAAAACGACGGCCAGT-3’ M13 reverse 5’-CAGGAAACAGCTATGACC-3’
Note: As most of the clones are heterozygous for the desired modification, multiple plasmids of each clone should be sent for sequencing since the pCR-Blunt plasmids will also contain amplicons for the unmodified allele.
- Prepare gDNA of every clone as described in step B1.
Data analysis
- The different analysis options for validation of the selected clones are straightforward assays typically resulting in non-ambiguous data. The PCR step after G418 selection of clones ensures the integration of the epitope tag and the selection cassette in the correct position of the genome. The PCR results should be positive for both homology arms to ascertain lack of aberrant recombination events. These clones are typically also analyzed by Southern blotting to confirm the presence of a single cassette. Random integrations can occur, leading to the presence of an additional construct in the selected PCR-positive clones. Cell clones showing extra bands on the Southern analysis should be eliminated for further use.
- After Cre-based removal of the selection cassette, PCR analysis spanning the modified site and the LOX scar should result in a band of the correct size. As mentioned above, PCR for clones wherein removal of the selection was not successful can fail due to size of the insert and the GC content of the promoter in the selection cassette. Clones with failed PCR reactions for the modified site should be eliminated for further analysis.
- Southern analysis further confirms removal of the cassette as the probe is directed against the Neomycin resistance gene. Clones showing multiple bands prior to TAT-Cre treatment are omitted for further validation, as these additional bands point to (a) random integration event(s). Selected Cre-treated clones with remaining bands after Southern blotting are not used for further analysis.
- Western analysis should confirm the presence of the epitope tag on the protein of interest. However, this implies that the protein is sufficiently expressed to allow detection by Western blotting. In some cases, a stimulus or a stabilizing agent (e.g., proteasome inhibitor) can be added to the cells to increase the levels of the protein (e.g., MDM2, Vandemoortele et al., 2016). Failure to detect the tagged protein by Western blotting is not necessarily an indication to exclude the cell line for further analysis but may complicate further downstream analysis. Note also that actual protein expression may vary between different clones. The testing of additional clones can still reveal clones with positive Western blot results. Note also that the size of the protein can deviate from the major protein isoform described in the database due to alternative splicing, protein processing, posttranslational modifications, incorrect annotation, etc. Size confirmation of the protein can be obtained with a specific antibody.
- Sequence verification is required to ensure correct integration of the tag and eliminates rare clones with frameshifts or other mutations that may have occurred during the engineering process.
- Only clones passing all previous assays are withheld for further gene-specific validation assays (such as modulation by a stimulus or protein interaction profiling).
Recipes
- Phase Lock lysis buffer
20 mM Tris-HCl, pH 7.5
5 mM EDTA
0.15 M NaCl
0.2% SDS
6 U proteinase K from Tritirachium album
Add ddH2O to 500 µl - TE-buffer
10 mM Tris-HCl, pH 7.5
1 mM EDTA - 2x HEBS (HEPES-buffered saline)
0.28 M NaCl
50 mM HEPES
1.5 mM Na2HPO4
800 ml ddH2O
Adjust to pH 7.05
Add ddH2O to 1 L
Note: The exact pH is critical for transfection efficiency. 2x HEBS can be stored at -20 °C. Avoid repeated freeze thaw cycles. - rAAV cell lysis buffer
0.15 M NaCl
50 mM Tris-HCl, pH 8.5
Filter sterilize using 0.22 μm filter - Direct PCR lysis buffer
2.1 ml Direct PCR lysis reagent (cell)
17 U proteinase K from Tritirachium album - CHAPS lysis buffer
50 mM HEPES, pH 7.4
100 mM NaCl
0.8% CHAPS (w/v)
cOmplete protease inhibitor - Depurination buffer
0.25 N HCl - Denaturation buffer
1.5 M NaCl
0.5 N NaOH - Neutralization buffer
0.5 M Tris, pH 7.4
1.5 M NaCl - 20x SSC
3 M NaCl
0.3 M sodium citrate tribasic dehydrate
Adjust to pH 7.0
Add ddH2O to 1 L
Sterilize - Wash buffer 1
200 ml 10x SSC
10 ml 10% SDS
790 ml ddH2O - Wash buffer 2
100 ml 10x SSC
10 ml 10% SDS
890 ml ddH2O - Wash buffer 3
10 ml 10x SSC
10 ml 10% SDS
980 ml ddH2O
Acknowledgments
G.V. is a PhD student supported by the Fund for Scientific Research – Flanders (FWO) (grant 3G050913N). S.E. acknowledges support from FWO (grants G011312N and 3G050913N). Cell lines constructed using this protocol were described in ‘An extra dimension in protein tagging by quantifying universal proteotypic peptides using targeted proteomics’ by Vandemoortele et al. (2016) and ‘Intelligent mixing of proteomes for elimination of false positives in affinity purification-mass spectrometry’ by Eyckerman et al. (2016), doi:10.1038/srep27220 and doi: 10.1021/acs.jproteome.6b00517 respectively.
References
- Chen, F., Pruett-Miller, S. M., Huang, Y., Gjoka, M., Duda, K., Taunton, J., Collingwood, T. N., Frodin, M. and Davis, G. D. (2011). High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods 8(9): 753-755.
- Eyckerman, S., Impens, F., Van Quickelberghe, E., Samyn, N., Vandemoortele, G., De Sutter, D., Tavernier, J. and Gevaert, K. (2016). Intelligent mixing of proteomes for elimination of false positives in affinity purification-mass spectrometry. J Proteome Res 15(10): 3929-3937.
- Khan, I. F., Hirata, R. K. and Russell, D. W. (2011). AAV-mediated gene targeting methods for human cells. Nat Protoc 6(4): 482-501.
- Ledford, H. (2015). CRISPR, the disruptor. Nature 522(7554): 20-24.
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J. and Gerard, G. F. (2004). Mutation detection using Surveyor nuclease. Biotechniques 36(4): 702-707.
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11): 2281-2308.
- Russell, D. W. and Hirata, R. K. (1998). Human gene targeting by viral vectors. Nat Genet 18(4): 325-330.
- Vandemoortele, G., Staes, A., Gonnelli, G., Samyn, N., De Sutter, D., Vandermarliere, E., Timmerman, E., Gevaert, K., Martens, L. and Eyckerman, S. (2016). An extra dimension in protein tagging by quantifying universal proteotypic peptides using targeted proteomics. Sci Rep 6: 27220.
- Vasileva, A., Linden, R. M. and Jessberger, R. (2006). Homologous recombination is required for AAV-mediated gene targeting. Nucleic Acids Res 34(11): 3345-3360.
- Wu, Z., Yang, H. and Colosi, P. (2010). Effect of genome size on AAV vector packaging. Mol Ther 18(1): 80-86.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Vandemoortele, G., De Sutter, D. and Eyckerman, S. (2017). Robust Generation of Knock-in Cell Lines Using CRISPR-Cas9 and rAAV-assisted Repair Template Delivery. Bio-protocol 7(7): e2211. DOI: 10.21769/BioProtoc.2211.
Category
Molecular Biology > DNA > Mutagenesis
Molecular Biology > DNA > DNA recombination
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.