- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitative Analysis of Exosome Secretion Rates of Single Cells
(*contributed equally to this work) Published: Vol 7, Iss 4, Feb 20, 2017 DOI: 10.21769/BioProtoc.2143 Views: 13495
Reviewed by: Gal HaimovichShalini Low-NamMarco Di Gioia
Original research article
The authors used this protocol in:

Jun 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
To study the inhomogeneity within a cell population including exosomes properties such as exosome secretion rate of cells and surface markers carried by exosomes, we need to quantify and characterize those exosomes secreted by each individual cell. Here we develop a method to collect and analyze exosomes secreted by an array of single cells using antibody-modified glass slides that are position-registered to each single cell. After each collection, antibody-conjugated quantum dots are used to label exosomes to allow counting and analysis of exosome surface proteins. Detailed studies of exosome properties related to cell behaviors such as responses to drugs and stress at single cell resolution can be found in the publication (Chiu et al., 2016).
Keywords: Single cellBackground
Exosomes have been found to play an essential role in tumorigenesis, cell-cell signaling, organotropic metastasis, drug resistance, and many crucial biological processes involving cell-cell communications. Most exosome isolation methods developed to date use ultracentrifugation at 100,000 x g (Théry et al., 2006) and require a large amount of samples. Combinations of microfluidics with immunological separation or physical trap have been reported (Liga et al., 2015) as simpler exosome isolation methods requiring a relatively small amount of sample. However, most microfluidic platforms have difficulties in integration of standard cell culture protocols, while cell culture in microfluidic environments can introduce new variables and unintended stresses to cells and change their behaviors and gene expressions. Above all, all existing approaches collect exosomes from cells without distinction, so it is extremely difficult to trace exosomes to the cells that secrete them. However, given the high diversity and inhomogeneity of biological samples, it is of great value to correlate the exosomes to the cell source. Furthermore, it is highly desirable to quantify the exosome analysis at a single cell level by finding the changes in exosome properties and secretion rates when cells are affected by stimuli, stresses and/or environmental changes. Here we provide a culture friendly, high-throughput, and versatile single-cell assay that enables quantitative analysis of exosomes secreted by individual cells.
Materials and Reagents
- 35 mm Petri dish (Corning, Falcon®, catalog number: 351008 )
- Cover glass (Ted Pella, catalog number: 260364 )
- Glass slides
- Silicon wafer (4” test grade) (University Wafer, catalog number: 452 )
- Acrylic (PMMA, 2 mm thick) (Sigma-Aldrich, catalog number: GF10188996 )
- 35 mm cell culture dish (Sigma-Aldrich, catalog number: D7804 )
- Parafilm (Sigma-Aldrich, catalog number: P7793 )
- Aluminum foil
- 0.22 µm filter
- Cells
Note: Our protocol can be used for both adherent and non-adherent cells. For examples, MCF7, MB-MDA-231, MCF10A, Neuronspheres, etc. - Polydimethylsiloxane (PDMS) (Dow Corning, catalog number: SYLGARD® 184 Silicone Elastomer Kit )
- Pure ethanol (Decon Labs, catalog number: V1001 )
- (3-mercaptopropyl)trimethoxysilane (MPS) (Gelest, catalog number: 4420-74-0 )
- Sulfo-GMBS, N-[γ-maleimidobutyryloxy]sulfosuccinimide ester (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 22324 )
- Phosphate-buffered salines (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023 )
- Monoclonal anti-human CD63 antibody (Ancell, catalog number: 215-820 )
- Bovine serum albumin (BSA power) (Sigma-Aldrich, catalog number: A8531 )
- Paraformaldehyde solution (4% in PBS) (Affymetrix, catalog number: 19943 1 LT )
- Biotinylated anti-CD63 (Ancell, catalog number: 215-030 )
- Qdot® incubation buffer (Thermo Fisher Scientific, Molecular ProbesTM, catalog number: Q20001MP )
- Quantum dots (Qdot® 545 ITKTM) streptavidin conjugate (Thermo Fisher Scientific, Molecular ProbesTM, catalog number: Q10091MP )
- Tris buffered saline with Tween® 20 (TBST-10x) (Cell Signaling Technology, catalog number: 9997 )
- DI water
- Blocking buffer (see Recipes)
- Blocking buffer for Qdot (see Recipes)
- 1x TBST (see Recipes)
Equipment
- CNC (Computer Numeric Control) micro milling machine (Minitech Machinery, model: Mini/Mill/1 )
- Disco automatic dicing saw 3220 (Nano3, model: 3220)
- Plasma Etch system (for cleaning) (Plasma Etch, model: PE-100 )
- Soda Lime Silica glass Petri dish (Corning, catalog number: 70165-152 )
- Standard biosafety cabinet
- Shaker (VWR, model: VWR Mini shaker )
- Oven
- 1 ml pipet
- Microscope (KEYENCE, model: BZ-9000 , similar to BZ-X700 )
- Centrifuge (Thermo Fisher Scientific, model: CL2 )
- Tweezers
- Cell counter
Notes
- Equipment 1-3 can be found in core facility such as micro/nano fab. Please see Nano3 facility of UCSD as an example. Equipment 1 can also be found in most machine shop as a service.
- Centrifuge is chosen to fit 35 mm dishes. If you have a centrifuge that suit 6-well plate, 35 mm dishes can be replaced with a 6-well plate.
Procedure
- Overall procedure for Single-cell assay used for analyzing exosome secretion is shown in Figure 1. Details for preparing each material will be described in step 2.
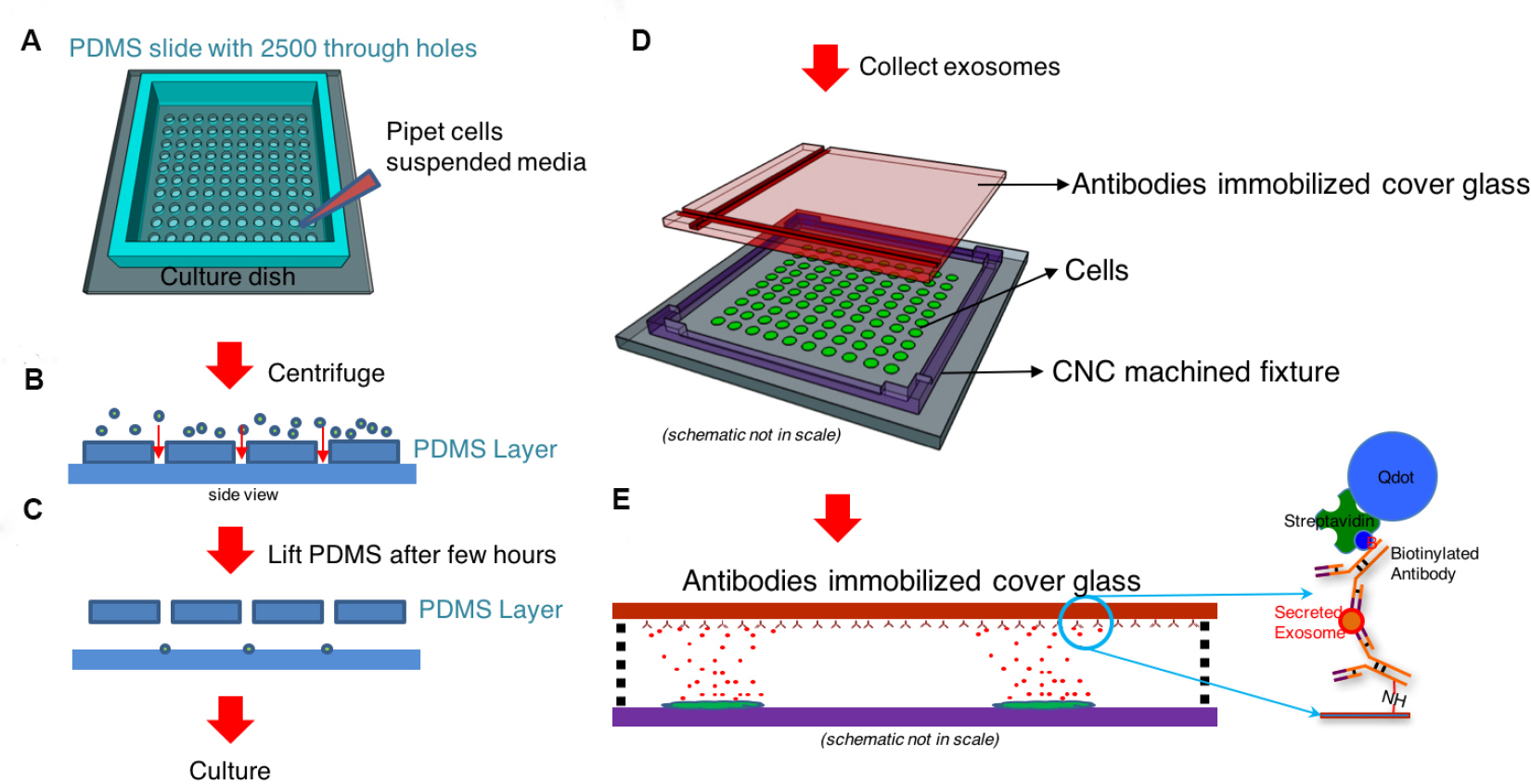
Figure 1. Workflow for single-cell assay used for analyzing exosome secretion. A and B. Loading single cells onto a culture dish with a cell loading chip that contains through hole arrays. C. The cell loading chip may be removed after cell attachment. D. A surface functionalized glass slide is placed in a CNC machined frame 100 µm above the cells. E. The glass slide with collected exosomes secreted by the corresponding cells. The captured exosomes are labeled with another biotinylated antibody and streptavidin-conjugated quantum dots to become visible under fluorescent microscope. - Prepare a cell loading chip that can fit into a 35 mm dish. A chip size between 5 x 5 mm2 and 12 x 12 mm2 is recommended. A cell loading chip made of PDMS is also recommended (Chiu et al., 2016).
- CNC machined fixture
- Design a fixture with an outer diameter fitted to a 35 mm dish. Inner square of the fixture is 50 μm greater (on each side) than the cover glass used for exosome collection (Figure 2).
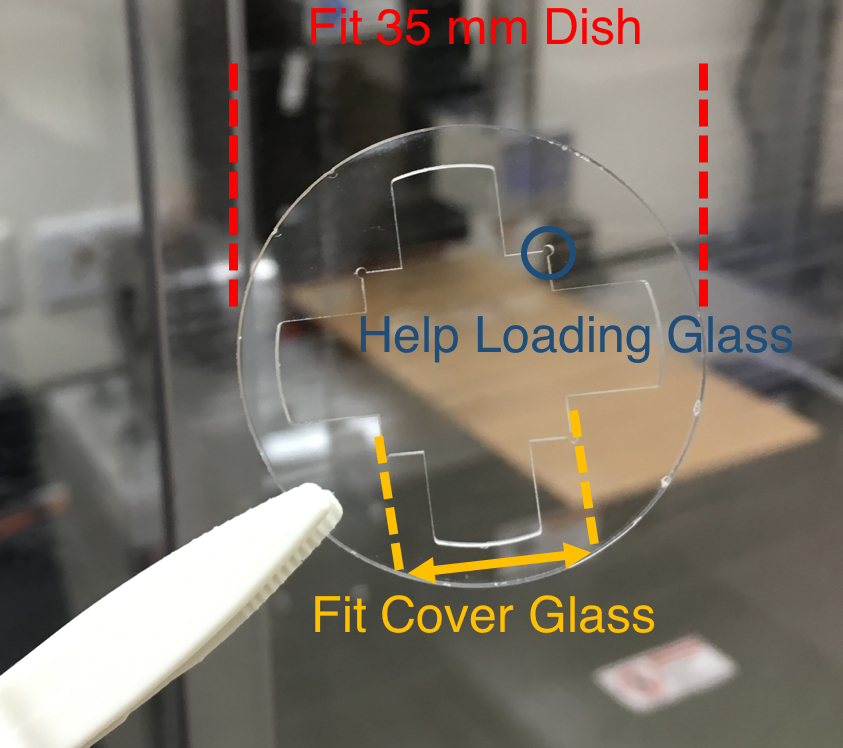
Figure 2. A machined PMMA that fit a 35 mm dish and exosome collection cover glass - Use CNC system to machine PMMA
- For adherent cell or cell loaded directly onto the dish, four diced silicon slides (100 μm x 3 mm) are attached (Note: we use PDMS to attach) to the inner corners as spacers between the cells and the collection glass (Figure 3).
- For non-adherent cell, the machined PMMA can be used after wash.
- Clean the machined PMMA using 75% ethanol and UV for 15 min.
Note: We use standard biosafety cabinet UV lamp.
Figure 3. Fixture with 4 100 µm spacers to keep the distance between the cover glass and cells
- Design a fixture with an outer diameter fitted to a 35 mm dish. Inner square of the fixture is 50 μm greater (on each side) than the cover glass used for exosome collection (Figure 2).
- Antibody immobilization on cover glass (this step will take about 4-6 h)
- Dice cover glass into 7 x 7 mm size.
- Using the dicing machine to cut ~30 μm depth fiducial on one side of cover glass. The design of fiducial is shown in Figure 4.

Figure 4. Cover glass with 30 μm depth fiducial - Clean the cover glass with O2 plasma (Plasma Etch System, PE100) at 200 W for 1.5 min on each side of cover glass.
- Silanization
- Prepare a sterile 35 mm glass dish.
Note: For silanization we used a glass dish rather than plastic dish. - Add 3 ml ethanol into the dish.
- Add 120 μl (3-mercaptopropyl)trimethoxysilane (MPS) into the dish (to get 4% v/v MPS) and mix well.
- Immerse cover glass in 4% MPS solution for 30 min on a shaker at 200 rpm.
Note: Flat surface without fiducial facing up. - Prepare a new 35 mm Petri dish with 3 ml ethanol.
- Transfer glass slides into the new dish and shake for 5 min at 200 rpm.
- Repeat steps 3d.v and 3d.vi two additional times.
- Transfer the glass slides into a new glass dish.
- Put the glass slides into a 100 °C oven and dry the slides for 30 min (until fully dry).
Note: Freshly used. Go to next step immediately. - Sulfo-GMBS coating
- Weigh 0.764 mg Sulfo-GMBS.
- Dissolve Sulfo-GMBS into 2 ml PBS to get 1 mM sulfo-GMBS solution in a 35 mm glass dish.
- Immerse the cover glass in the solution.
- Put the glass dish on the shaker at 200 rpm and incubate for 10 min and another 30 min without shaking.
- Rinse with PBS (3 ml) for 5 min 3 times on the shaker at 200 rpm.
Note: Here we use plastic dish for PBS washing. - Anti-CD63 Ab immobilization
- Prepare a 35 mm Petri dish.
- Add 2 ml PBS into the dish and shake gently to wet the whole surface.
- Add 5 μg (5 μl) anti-CD63 Ab into the dish (~0.05 μM).
- Use 1 ml pipet to mix the solution 20 times.
- Immerse the cover glass into the anti-CD63 Ab solution.
- Incubate the dish at 4 °C for 2 h.
- Rinse the dish with PBS (3 ml) for 5 min 3 times on the shaker at 200 rpm.
- Block Non-reacted GMBS
- Pipet 3 ml 5% bovine serum albumin (BSA) solution into a sterile 35 mm dish.
- Immerse the glass into BSA solution for 30 min at 4 °C.
- Pipet 3 ml 5% bovine serum albumin (BSA) solution into a sterile 35 mm dish.
- Storage
- Immerse the treated cover glass in PBS at 4 °C.
Notes:
1) There is no need to wash with PBS after BSA blocking.
2) The treated cover glass can be stored for at least 2 months. (We have never tried longer than this duration.)
- Immerse the treated cover glass in PBS at 4 °C.
- Dice cover glass into 7 x 7 mm size.
- Single-cell loading
- Adhere cell loading array (through hole PDMS for adherent cell or PDMS wells for non-adherent cells) at the center of 35 mm cell culture dish. Figure 5 is an example of through hole PDMS adhere to a 35 mm dish.

Figure 5. Through hole PDMS adhere to a 35 mm dish - Place the CNC machined fixture on top of cell loading array.
Note: For non-adherent cells, loading the cells after placing the fixture; for adherent cells, placing the fixture after loading and peel off the PDMS mesh. - If the fixture is designed to fit the inner diameter of 35 mm dish, it can be placed directly on top of PDMS.
- If the fixture is not designed to fit the inner diameter of 35 mm dish, one would need to use some uncured PDMS to seal the side of fixture to the cell loading array. Make sure PDMS is fully cured before use.
- Use O2 plasma to treat the whole dish (with cap open) for 30 sec at 200 W. This will make PDMS more hydrophilic.
- Sterilize the whole dish under UV (15 W) for 15 min.
- Dispense 2-3 ml culture media into the dish and store in 37 °C overnight (at least 4-6 h depends on the well size). This step will help remove small bubbles trapped in PDMS wells, and ensure no bubbles after ramping up temperature.
- Before harvesting the cells, check under microscope and make sure there are no bubbles in the PDMS wells.
- Harvest the cell based on normal procedure.
- Dilute the cells with culture media for a suitable density. For a 50 x 50 array of loading wells with size of 40-50 μm and 250 μm separation (center to center), about 50,000-100,000 cells per ml will give good number of single cells.
- Pipet diluted cells into the dish, and homogenize the cells by pipetting the diluted cells a few times.
- Seal the dish with Parafilm.
- Put the dish in the centrifuge and use tape of cap to secure the dish. Two dishes each time will help to balance centrifuge easily (Video 1).
 Video 1. Loading cells by centrifuge
Video 1. Loading cells by centrifuge - Centrifuge for 1 min at 140 x g.
- After centrifuging, check the loading condition under microscope (2x or 4x of objective lens). You will see single/double cells cover more than 75% of wells. If loading efficiency is too low, one can repeat steps 4i-4l.
- Clear unwanted cell
- For adherent cells, let the cells settle for 6-8 h before removing the PDMS mesh. After removal, the unwanted cells will be taken away with the PDMS mesh. Wash the whole plate with media for 3 times.
- For non-adherent cells, after centrifuge, tilt the dish to 45-60 degree and use pipet to flush the surface. Repeat this step with clean media for 2-3 times.
- Exosome collection
Note: Steps 6, 7, and 8 will take about 10 h including 3 h of exosome collection. We recommend finish 6-8 in one day without interruption. - To start the collection process, aspirate the media in the dish and rinse with exosome free media or PBS.
- Dispense exosome free media into the dish.
- Carefully take out the treated cover glass from step 3 (Video 2). Video 2. Place the treated cover glass on top of the cells loading array
- Flip the cover glass. Let the flat surface without fiducial face down. Put the glass slide in the CNC machined PMMA fixture (Video 2).
- Put the cells back to the incubator and wait for 1-3 h based on the experimental plan.
- Image sample and exosome fixation
- After the exosome collection, capture whole dish images with microscope. Use the fiducial to find the focal plane.
- Prepare a 35 mm Petri dish filled with 2 ml of 4% paraformaldehyde in PBS.
- Use tweezers to take out the cover glass.
- Flip the cover glass and let the flat side (without fiducial) face up, and immerse the slide immediately to the paraformaldehyde for 30 min at room temperature.
Note: Don’t forget to flip the cover glass. - Label Qdots
- After exosome fixation, wash with 1x PBS for 3 times, each time 5 min at 200 rpm.
- Blocking
- Immerse the cover glass in 5% BSA for 2 h at room temperature.
- After blocking, wash with 1x PBS for 3 times, each time 5 min at 200 rpm.
- Label Biotinylated anti-CD63 Ab or other antibody
- Prepare a 35 mm Petri dish.
- Create a solution of 800 μl of PBS, 200 μl of 5% BSA, and 2.5 μl of Biotinylated anti-CD63 Ab.
- Immerse the cover glass in the solution for 2 h at 4 °C.
- Wash with 1x PBS for 3 times, each time 5 min at 200 rpm.
- Blocking
- Prepare a 5% BSA in Qdots incubation buffer.
- Dispense 5% BSA in Qdots incubation buffer into a 35 mm Petri dish.
Note: It is recommended to use 500 μl for a 7 x 7 mm-12 x 12 mm glass slides to save the cost of Qdots. Add the blocking buffer from the side of dish till the buffer covers the glass. - Immerse glass slide in the buffer for 2 h at room temperature.
- Qdots labeling
- Mix well 1 ml Qdots incubation buffer and 10 μl Qdots to get 10 nM Qdots solution.
- Immerse the cover glass in the solution for 1 h at room temperature. Cover the dish with aluminum foil.
Note: No washing process before this step. - Washing
- Preheat 1x TBST buffer and DI water at 50 °C for 30 min before Qdots labeling finishing.
- Add 2 ml 50 °C 1x TBST into a new dish, and transfer the cover glass to the dish with 1x TBST solution.
- Put the dish on the shaker and wash for 5 min at 200 rpm.
- Repeat steps 7f.ii-7f.iii for three times.
- Add 2 ml 50 °C deionized water (DI water) into another dish, and transfer the glass slide into it.
- Put the dish on the shaker and wash for 5 min at 200 rpm.
- Repeat steps 7f.v-7f.vi for three times.
- Dehydration
- Dispense 2 ml of 30% ethanol, 50% ethanol, 75% ethanol, 90% ethanol, and two portions of 100% ethanol in each clean dish.
Note: The serial graded ethanol solutions are prepared with sterilized ddH2O. - Immerse the washed sample in 30% ethanol for 30 sec, 50% ethanol for 30 sec, 75% ethanol for 30 sec, 90% ethanol for 30 sec, and 2 dishes of 100% ethanol for 30 sec.
- Transfer the glass on a clean surface and let it air dry. Cover with aluminum foil during drying.
- Imaging and counting
- Register fiducial on exosome collected glass slide relative to the position of cell array.
- Open the image taken at step 6 that contains fiducial, e.g., cross pattern.
- Display the image on computer screen using 4x microscope objective lens.
- Cover the computer screen with a piece of transparency (Figure 6).
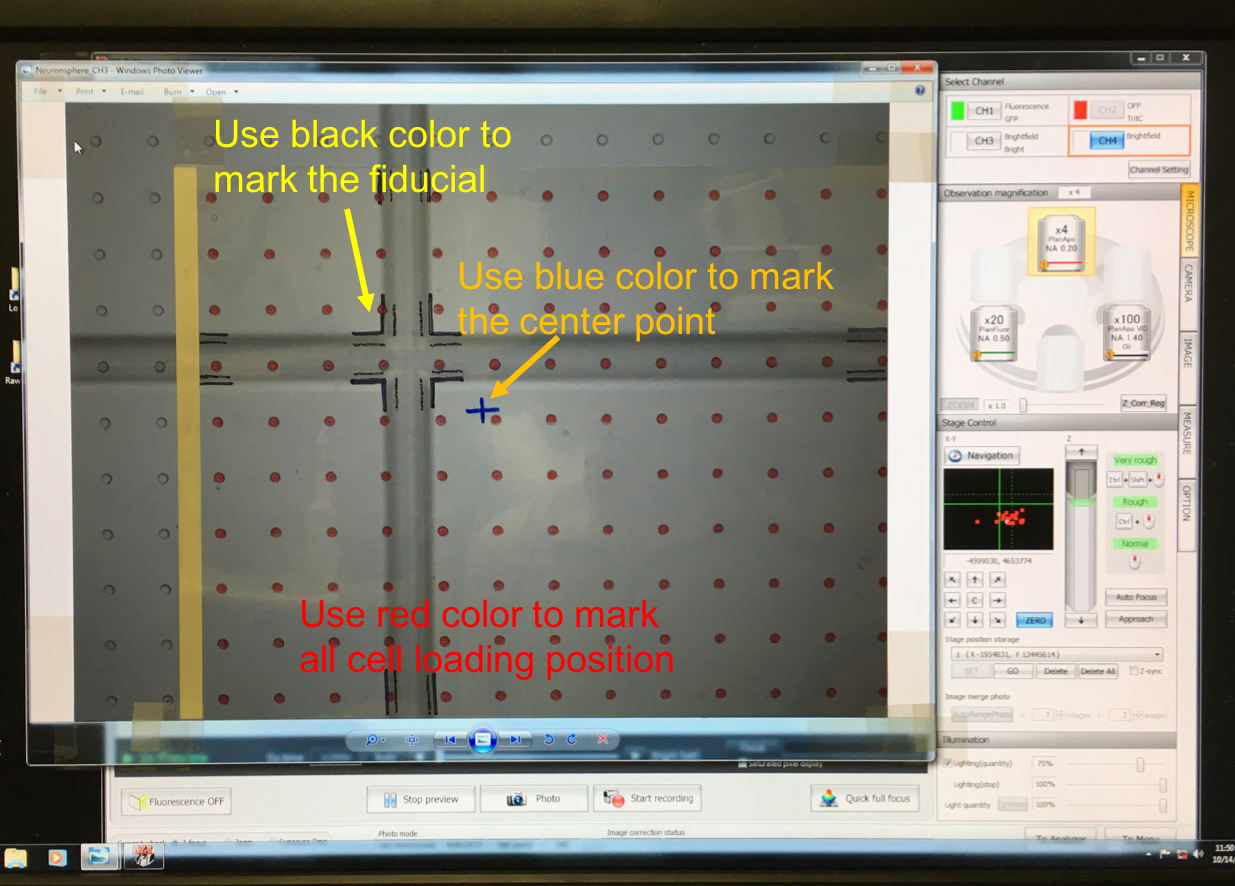
Figure 6. Transparency with labeled cell positions (red dots) and fiducial (cross pattern) - Use a black marker to trace the edge of fiducial on the transparency.
- Use a red marker to draw the cell sites on the transparency.
- One transparency can cover ~5 x 6 mm2 area on glass. Use another transparency on another cross fiducial position if the size of cell array is greater than the field of view of 4x objective.
- Place glass slide under an inverted microscope with the exosome side facing down (i.e., facing the objective lens of microscope). We use double-sided tape to adhesive the edge of the cover glass on a glass slide.
Note: Double-sided tape will contribute a lot fluorescent signal. Do not overlap the tape with the sample area. - Register exosomes collection sites/cell loading sites
- At this step, we will register exosome collection sites on the glass slide to the center of the field of view initially using 2x/4x objective lens and then 100x objective lens to count the quantum dots that label the exosomes.
- Adhere the marked transparency on the screen (Figure 7A).
- Switch to 2x/4x objective lens or any low magnification.
- Move the sample stage until the fiducial on the glass slide is aligned with the fiducial drawn on the transparency (Figure 7B).
- Mark center position on the transparency. This will become the center field under 100x objective.
- Choose one cell loading site (Cell-1). Adjust the sample stage to move the chosen cell loading site to the center position (marked as Center in the Figure 7C). Record micrometer readings of the sample stage at this position as position-1.
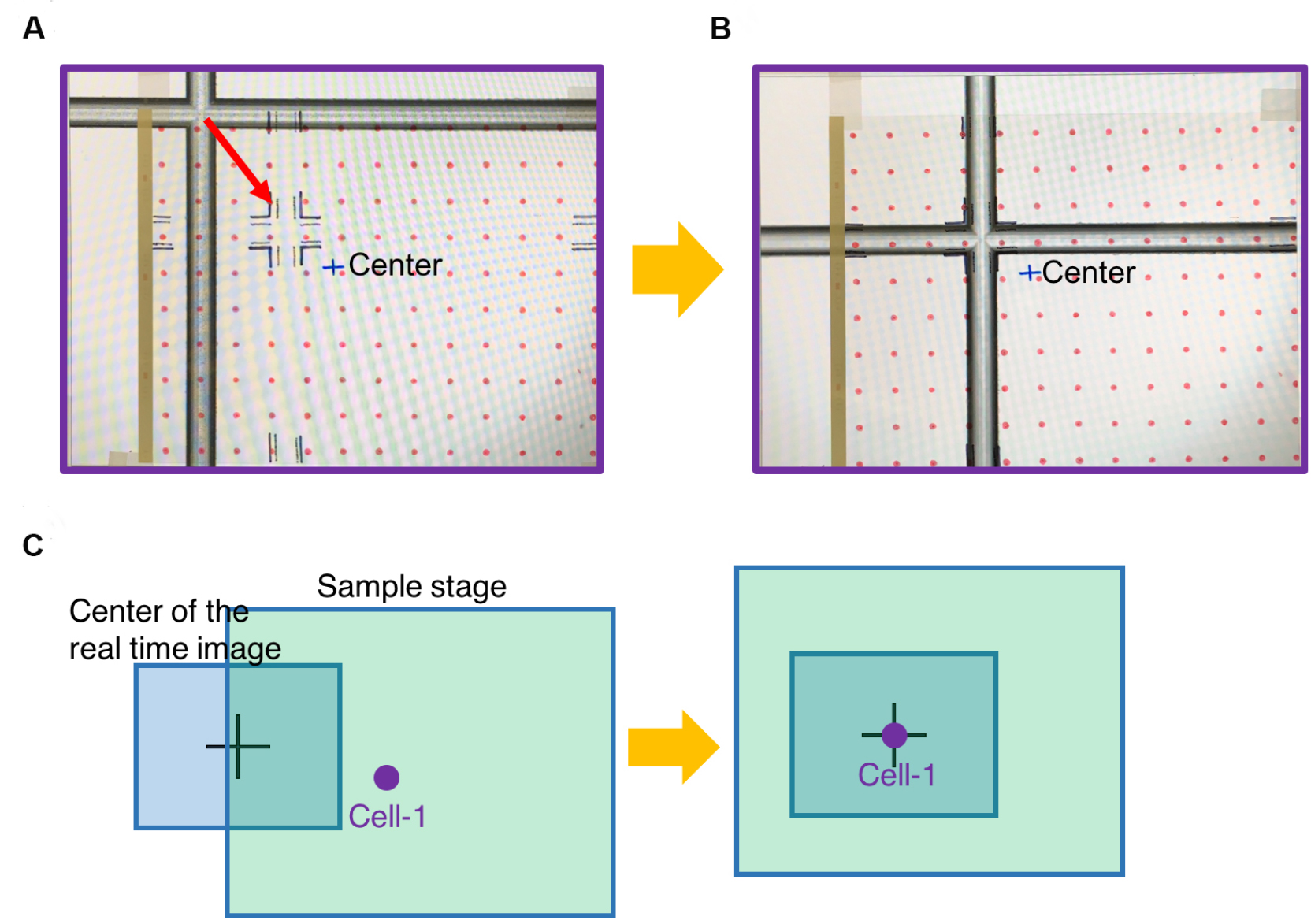
Figure 7. Register the first cell loading site. A. Viewing the image of glass slide on the computer screen covered by the marked transparency. B. Move the glass slide to align the fiducial with the marked position on the transparency. C. Move the chosen cell loading site (Cell-1) to the center position of the transparency, which is in the center field of 100x objective. - To register the rest of exosome collection sites, simply count how many spots (red circles) the chosen site is away from the first cell (Cell-1). Under 2x/4x objective lens, move the micrometers of the sample stage according to the red circles on the transparency. This will move the exosome collection site to the center of image (Figure 8).
- Register all the exosome collection sites of interest by repeating step 9c.vii.
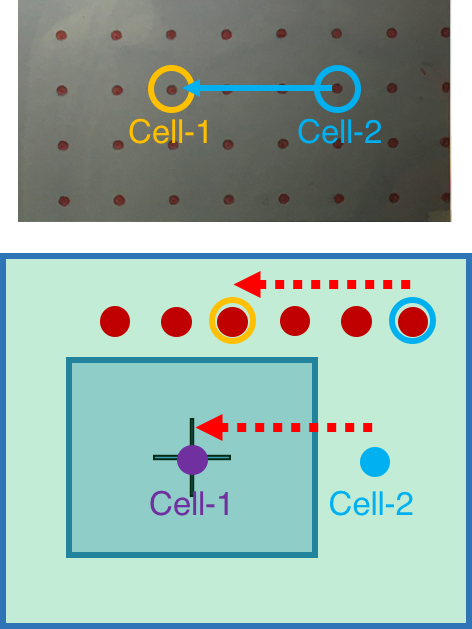
Figure 8. Register the rest of cell positions
- At this step, we will register exosome collection sites on the glass slide to the center of the field of view initially using 2x/4x objective lens and then 100x objective lens to count the quantum dots that label the exosomes.
- Imaging
- Switch to 100x objective lens
- Apply real time haze reduction and random noise cancellation if applicable. This will help to identify Qdot signals from the noise signals generated due to long exposure time. Figure 9A shows an example of an image after haze reduction.
- Increase exposure time to 1.5-2.5 sec. It is easier to distinguish Qdots from random noise while using long exposure time. Although you may see both on the monitor, Qdots are always brighter than the noise.
- Adjust the focus slowly to see the Qdot signals.
- Optimize the focus carefully for the highest Qdot image contrast.
- Take image with a long exposure time, e.g., 2.5 sec is recommended.
- Qdots counting
- Black balance. Select an area and adjust the black balance until the dimmer Qdots show on the screen. Figure 9 shows an example noise reduction after black balance.
Note: We suggest showing the image as original size while adjusting black balance. Some Qdots may not be visible due to the setting or capability of the computer screen. - Use cell counter to count the illuminated dots. Carefully set the threshold.
- Repeat steps 8f.i and 8f.ii to count all the images.
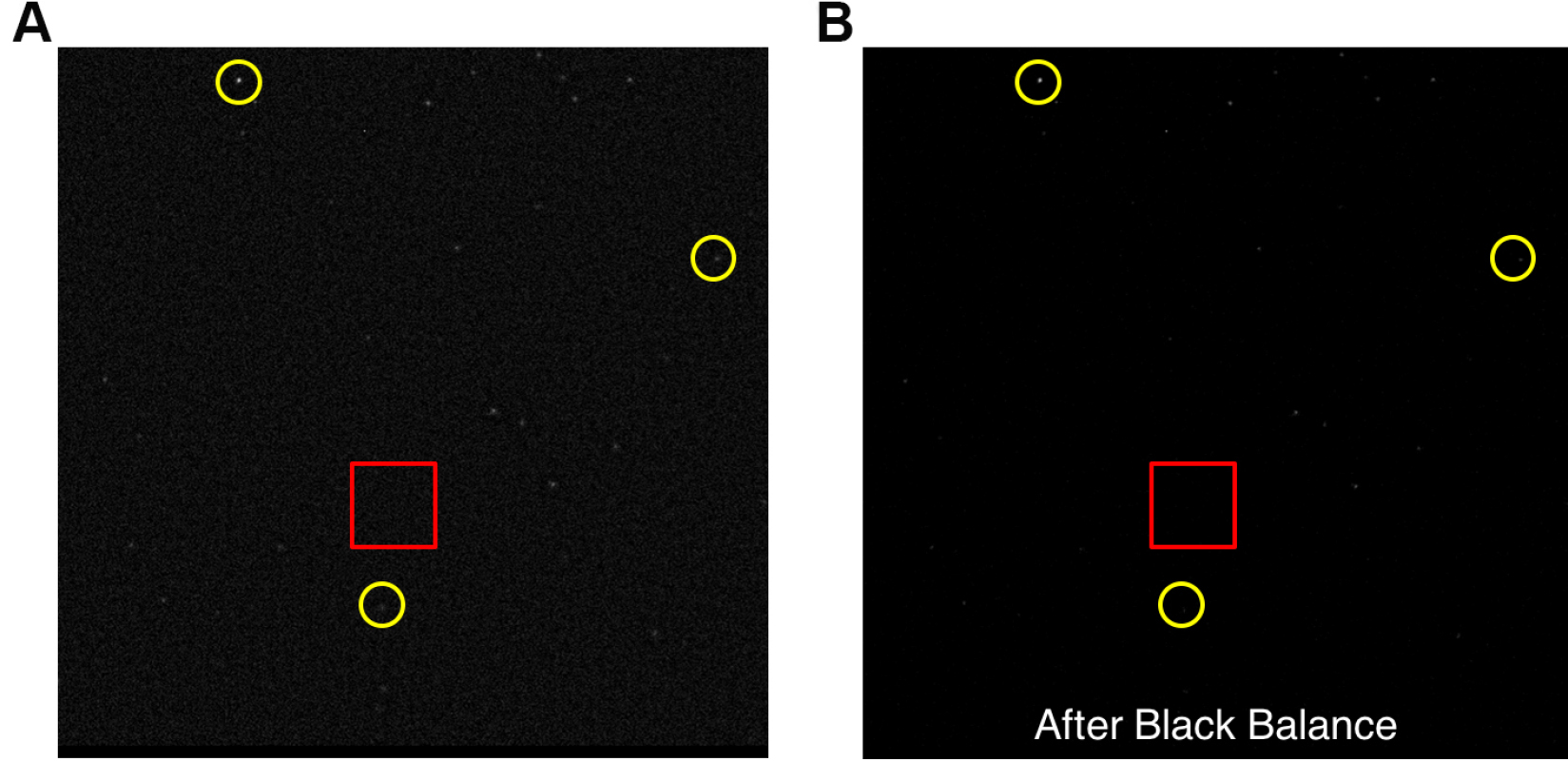
Figure 9. An example of Qdots image. A. Image after haze reduction and random noise cancellation. Red rectangular area shows background noise under very long exposure time. Yellow circles are examples of Qdots with different intensities. B. Image after black balance.
Data analysis
![]()
Where,
R: Single cell exosome secretion rate,
N: Number of Q-dots in the exosome collection slide area corresponding to the cell position,
Nn: Noise level,
C: Cell number in one site,
T: Exosome collection time.
We only imaged the sites that contain a single cell initially. If the cell divided during the experiment, we assumed the same exosome secretion rate for all the cells in the site.
Notes
- Washing: We use clean 35 mm Petri dish at every washing step.
- At step 8c, different antibodies can be chose to target the exosomes depending on the purpose of study. Here we use anti-CD63 antibody just as an example.
- At step 8e Imaging, if it is hard to find Qdots plane, one can follow the steps here:
- Under lower magnification (2x or 4x), move the fiducial to the center of the view.
- Switch to 100x magnification. The fiducial should still be seen within the field of view. If not, adjust the sample stage till the fiducial pattern shows in the field of view. As shown in Figure 10, try to focus on plane-1.
- Keep lowering the lens (i.e., bringing the lens away from the glass slide) until the lowest plane of fiducial is in focus marked as plane-2 in Figure 10. Now one can be sure the microscope is imaging a z-position between the top surface and the bottom surface of the glass slide.
- Keep lowering the lens, the Qdots and exosomes should be at the plane-3 shown in Figure 8.
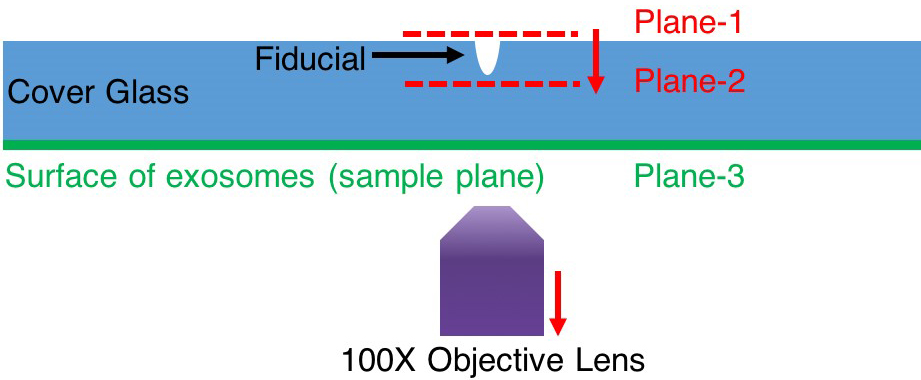
Figure 10. Find focal plane of exosomes
- Under lower magnification (2x or 4x), move the fiducial to the center of the view.
Recipes
- Blocking buffer
- Dissolve 0.5 g BSA into 25 ml PBS
- Filter the solution with 0.22 µm filter
- Store the 5% BSA buffer at 4 °C
- Blocking buffer for Qdot
- Dissolve 0.5 g BSA into 25 ml Qdots buffer
- Filter the solution with 0.22 µm filter
- Store the 5% BSA Qdots buffer at 4 °C
- 1x TBST
Diluted 10x TBST by DI water
Acknowledgments
We acknowledge the article of journal publication on Small 2016, 12 (27), page 3658-3666. This protocol is modified from the experimental section of this Small article. The authors acknowledge the technical support of the staff of the San Diego Nanotechnology Infrastructure (SDNI), which is part of the National Nanotechnology Coordinated Infrastructure (NNCI). Research reported in this publication was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number R21GM107977 and the National Institute of Biomedical Imaging and Bioengineering (NIBIB) of the National Institutes of Health under Award Number R43EB021129. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Yu-Hwa Lo has an equity interest in Nanocellect, Inc., a company that may potentially benefit from the research results. He is a co-founder of the company and a member of the company’s Scientific and Advisory Board.
References
- Chiu, Y. J., Cai, W., Shih, Y. R., Lian, I. and Lo, Y. H. (2016). A single-cell assay for time lapse studies of exosome secretion and cell behaviors. Small 12(27): 3658-3666.
- Liga, A., Vliegenthart, A. D., Oosthuyzen, W., Dear, J. W. and Kersaudy-Kerhoas, M. (2015). Exosome isolation: a microfluidic road-map. Lab Chip 15(11): 2388-2394.
- Théry, C., Amigorena, S., Raposo, G. and Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3: Unit 3 22.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chiu, Y., Cai, W., Lee, T., Kraimer, J. and Lo, Y. (2017). Quantitative Analysis of Exosome Secretion Rates of Single Cells. Bio-protocol 7(4): e2143. DOI: 10.21769/BioProtoc.2143.
Category
Cell Biology > Organelle isolation > Exosomes
Cell Biology > Single cell analysis > Microfluidics
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.