- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

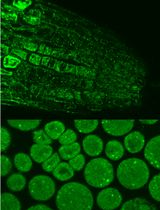
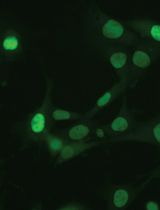
Immunofluorescence Analysis of Yeast Protein

Published: Vol 2, Iss 13, Jul 5, 2012 DOI: 10.21769/BioProtoc.211 Views: 17596
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Many important regulatory proteins such as transcription factors are regulated through subcellular localization. Protein localization can be examined by fusing a GFP tag. However, GFP is relatively big in size, and potentially may affect correct protein localization. Several small tags have been developed, such as myc, HA or Flag. By using immunostain and fluorescence microscopy as described in this protocol, one can easily probe the regulation of a selected yeast protein with the application of the aforementioned small tags.
Keywords: YeastMaterials and Reagents
- Yeast cells
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P8416 )
- Potassium phosphate dibasic (K2HPO4·3H2O) (Sigma-Aldrich, catalog number: P9666 )
- Sorbitol (C6H14O6) (Sigma-Aldrich, catalog number: S1876 )
- BSA (Albumin from bovine serum) (Sigma-Aldrich, catalog number: A4503 )
- Potassium chloride (KCl) (Thermo Fisher Scientific, catalog number: BP366-1 )
- 37% formaldehyde solution (Thermo Fisher Scientific, catalog number: F75P1GAL )
- Zymolyase (USB, catalog number: Z1001 )
- Vectorshield
- Poly-L-lysine
- DAPI
- Cytoseal 60
- Phosphate buffer (see Recipes)
- Sorbitol buffer (see Recipes)
- Blocking buffer (see Recipes)
Equipment
- Centrifuges
- Shaker
- Conical tube
- Fluorescence microscope
- 15 ml conical tube
- Light microscope
- Heat blot
Procedure
- Inoculate yeast cells overnight in a 30 °C shaker.
- Subculture cells at OD600=0.1 in 5 ml YPD or defined media.
- Continue to shake at 30 °C for 4-6 h to a concentration of 1-5 x 107.
- Add 0.6 ml of 37% formaldehyde solution directly to the cells and continue to incubate with shaking for 90 min (to be exact) at the same temperature as growth.
- Transfer cells to a 15 ml conical tube and pellet by centrifugation at 660 x g for 3 min at 4 °C.
- Aspirate supernatant and wash cells with ice-cold 5 ml phosphate buffer. Be gentle at this step because yeast cells are very fragile after fixation by formaldehyde.
- Repellet cells and wash with ice-cold 5 ml sorbitol buffer.
- Pellet cells and aspirate supernatant. Resuspend in ~1 ml sorbitol buffer.
- Pre-warm the cells at 30 °C for 5 min.
- Add 25 μl of zymolyase, mix gently and incubate on 30 °C heat blot for 15-30 min.
- Check the digestion under a light microscope (fully digested cells are gray while undigested cells are bright).
- When 80% of cells are digested, Pellet cells 660 x g for 3 min at 4 °C. Re-suspend cells gently in 0.5 ml ice-cold sorbitol buffer.
- Repeat step 12.
- Prepare slides by coating them with Poly-L-lysine for 10 min at room temperature (RT).
- Wash slides 5 times with water and let them dry at RT.
- Place 20 μl of cell suspension into each well on the slide. Incubate in a wet chamber for at least 10 min.
- Immediately immerse slide in ice-cold methanol for 6-7 min.
- Remove and immerse immediately in ice-cold acetone for 30 sec.
- Dry the slides on 30 °C heat blot.
- From this step on, don’t let the wells dry. Wash wells several times with blocking buffer and incubate a moist chamber for at least 10 min.
Note: longer incubation time may give better results, for example, incubate in blocking buffer at 4 °C overnight. - Remove supernatant and add 20 μl of primary antibody (diluted in blocking buffer). Incubate in a moist chamber for at least 2 h. Note: can put the slides at 4 °C overnight.
- Aspirate excess solution and wash 5x with blocking buffer.
- Add 20 µl of fluorescence conjugated-secondary antibody (diluted in blocking buffer).
- Place in a dark/moist chamber for ~1 h. Remember to keep slides in the dark as much as possible to prevent bleaching of fluorescence.
- Aspirate excess solution and wash 5x with blocking buffer.
- Then, wash once with 1x PBS.
- Dilute DAPI (1:1,000) in 1x PBS.
- Add DAPI solution to wells for 2 min.
- Aspirate excess solution and wash once with 1x PBS. Do not allow the slide to dry and add 2 µl Vectorshield to the well and immediately cover with cover slide.
- Seal the edge of the slides with Cytoseal 60 and examine under a fluorescence microscope.
Recipes
- Phosphate buffer
Make 0.1 M KH2PO4 in H2O
Make 0.1 M K2HPO4 in H2O
Add K2HPO4 solution to KH2PO4 solution to bring the pH to 6.5. - Sorbitol buffer
0.1 M potassium phosphate buffer (pH 6.5)
1.2 M sorbitol - Blocking buffer
5 % BSA in 1x PBS (pH 8.0)
Acknowledgments
This protocol was adapted from and used in Wei and Zheng (2009) and Wei et al. (2009).
References
- Wei, Y. and Zheng, X. F. (2009). Sch9 partially mediates TORC1 signaling to control ribosomal RNA synthesis. Cell Cycle 8(24): 4085-4090.
- Wei, Y., Tsang, C. K. and Zheng, X. F. (2009). Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J 28(15): 2220-2230.
Article Information
Copyright
© 2012 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wei, Y. (2012). Immunofluorescence Analysis of Yeast Protein. Bio-protocol 2(13): e211. DOI: 10.21769/BioProtoc.211.
Category
Microbiology > Microbial biochemistry > Protein > Immunodetection
Biochemistry > Protein > Fluorescence
Biochemistry > Protein > Immunodetection > Immunostaining
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.