- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Sequencing of Ebola Virus Genomes Using Nanopore Technology

Published: Vol 6, Iss 21, Nov 5, 2016 DOI: 10.21769/BioProtoc.1998 Views: 11752
Reviewed by: Yannick DebingEmily CopeEmilie Battivelli
Original research article
The authors used this protocol in:

Feb 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Sequencing of virus genomes during disease outbreaks can provide valuable information for diagnostics, epidemiology, and evaluation of potential countermeasures. However, particularly in remote areas logistical and technical challenges can be significant. Nanopore sequencing provides an alternative to classical Sanger and next-generation sequencing methods, and was successfully used under outbreak conditions (Hoenen et al., 2016; Quick et al., 2016). Here we describe a protocol used for sequencing of Ebola virus under outbreak conditions using Nanopore technology, which we successfully implemented at the CDC/NIH diagnostic laboratory (de Wit et al., 2016) located at the ELWA-3 Ebola virus Treatment Unit in Monrovia, Liberia, during the recent Ebola virus outbreak in West Africa.
Background
Determining the full-length sequence of virus genomes is an essential procedure in virology. While the classical approach to this involved Sanger sequencing following a primer-walking strategy, newer approaches involve the use of next-generation sequencing methods such as 454, Illumina or Ion Torrent technologies. A common problem with all these technologies, despite their many advantages, is that the required instrumentation is large, expensive, fragile, and therefore difficult to transport. Also, library preparation procedures are often involved. While under usual circumstances these issues are of little consequence, as these machines are run in specialized laboratories with excellent infrastructure, during virus outbreaks in remote areas (for example in case of ebolavirus outbreaks) this can pose significant problems, particularly since the export of samples from affected areas to these specialized laboratories is often politically and logistically challenging. Under these circumstances, the availability of a sequencing technology that can be easily and quickly deployed into remote areas, and allows sequencing to be done directly in an outbreak area, can be invaluable. Therefore, we have tested the MinION sequencing device, which at that time was under development by Oxford Nanopore Technologies (ONT), at the field diagnostic laboratory at the ELWA-3 Ebola virus Treatment Unit in Monrovia (de Wit et al., 2016) during the recent Ebola virus outbreak in West Africa, and developed a protocol for the rapid generation of full-length sequences of Ebola viruses under these conditions. This device employs nanopores, through which nucleotide-strands are transported in a controlled fashion. The nucleotides block and thus modulate an ion-current flowing through those pores, depending on the physical properties of the nucleotides passing through the nanopores, and these current modulations are measured by the device and translated into nucleotide sequences. Results of this test, which indicated that this technology indeed shows great promise as a rapidly deployable and highly usable sequencing platform, are available elsewhere (Hoenen et al., 2016), as are the results of a similar test using the same sequencing platform performed independently of our own efforts by Quick et al. (2016).
Materials and Reagents
Note: This protocol was established and tested during the Ebola virus outbreak in West Africa in January 2015, using materials and reagents available at that time. As the development of the MinION platform progresses rapidly, some modifications might be necessary to adopt the protocol to the materials and reagents available now. Particularly, while at the time the technology was only available to members of the MinION access program, it is now commercially available.
- 0.2 ml PCR-tubes (ideally in strips of 8) (e.g., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: AB0490 )
- 1.5 ml tubes (e.g., Thermo Fisher Scientific, Fisher Scientific, catalog number: S348903 )
- Eppendorf protein LoBind tubes, 1.5 ml, PCR clean (Eppendorf, catalog number: 0030108116 )
- Gloves
- MinION flowcell, revision 7.3 (Oxford Nanopore Technologies [ONT])
- RNA freshly purified (or stored at -80 °C) from patient blood samples following appropriate safety protocols
- SuperScript III First-Strand-Synthesis System (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18080051 )
- DEPC-treated nuclease-free water (e.g., Thermo Fisher Scientific, AmbionTM, catalog number: AM9906 )
- Primer, 10 μM (see Table 1 for sequences); primer CGGACACACAAAAAGAAAGAAG at a concentration of 2 μM and 10 μM
- dNTP mix, 10 mM each (e.g., New England Biolabs, catalog number: N0447S )
- iProofTM high-fidelity polymerase (Bio-Rad Laboratories, catalog number: 1725301 )
- Agencourt AMPure XP beads (Beckman Coulter, catalog number: A63881 )
- His-Tag dynabeads (Thermo Fisher Scientific, NovexTM, catalog number: 10103D )
- Magnetic stand for PCR-purification with Agencourt AMPure XP beads in 1.5 ml tubes (e.g., Beckman Coulter, catalog number: A29182 )
- 70% ethanol
- Qiagen elution buffer (QIAGEN, catalog number: 19086 )
- 50x TAE buffer (ideally in 10.2 ml aliquots) (e.g., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: B49 )*
- Agarose (ideally in pre-weighed aliquots of 0.5 g) (e.g., Thermo Fisher Scientific, InvitrogenTM, catalog number: 16500100 )*
- DNA standard (e.g., New England Biolabs, catalog number: N3200S )*
- 6x gel loading dye (e.g., New England Biolabs, catalog number: B7022S )*
- Fast Blast DNA stain (Bio-Rad Laboratories, catalog number: 1660420 )*
- NEBNext dA-tailing module (New England Biolabs, catalog number: E6053S )
- NEBNext end repair module (New England Biolabs, catalog number: E6050S )
- Blunt/TA ligase master mix (New England Biolabs, catalog number: M0367L )
- Genomic DNA Sequencing Kit SQK-MAP004 (ONT): contains DNA CS, 2x wash buffer, elution buffer, EP buffer, HP adapter, Adapter mix and Fuel mix
*Note: Optional materials and reagents.
Equipment
- MinION sequencing device (ONT)
- PCR cycler (e.g., Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: A24811 )
- Vortexer (e.g., Thermo Fisher Scientific, Fisher Scientific, catalog number: S96461A )
- Mini centrifuges for 1.5 and 0.2 ml tubes (e.g., VWR, catalog number: 93000-204 )
- Pipettes: 10 μl, 20 μl, 100 μl, 1,000 μl, 8 x 20 μl multichannel with filter tips (e.g., Mettler-Toledo, catalog numbers: PR-10 , PR-20 , PR-100 , PR-1000 , L8-20XLS+ )
- Racks for 0.2 ml PCR tubes and 1.5 ml tubes (e.g., Thermo Fisher Scientific, Fisher Scientific, catalog numbers: 05-541-85 , 05-541-2 )
- PCR cooler rack, 0.2 ml, 4 °C (e.g., Eppendorf, catalog number: 3881000031 )
- Styrofoam box with lid and cold packs (frozen cold pack at bottom, 4 °C cold pack on top, then sample rack on top of that) as cooler box (or alternatively ice box, when ice is available)
- Electrophoresis chamber with power supply (e.g., Mupid-exU, Takara Clontech)*
- Microwave oven*
- Agarose gel casting stand, or alternatively laboratory tape*
- 250-300 ml glass flask for preparing agarose*
- Box for agarose gel staining*
- Additional infrastructure when sequencing under outbreak conditions
Note: This infrastructure is a given in any Western laboratory, but should be considered when establishing the method under field conditions in outbreak areas. While this topic cannot be exhaustively covered in this protocol, the following points should be carefully considered.- Power generator, uninterruptable power supplies, voltage regulators (all depending on the quality of the electricity supply).
- Air conditioning if possible, alternatively external heatsink (e.g., a ~30 x 30 cm metal plate) for MinION sequencing device.
- Clean water for preparation of 1x TAE, and for DNA staining and destaining - bottled drinking water (ideally in 500 ml bottles) can be used for this purpose if no other source of clean water is available.
- Equipment for safe sample inactivation and RNA extraction.
- 4 °C fridge, -20 °C freezer, optionally -80 °C freezer for storage of RNA.
- Personal protective equipment as deemed necessary.
- Power generator, uninterruptable power supplies, voltage regulators (all depending on the quality of the electricity supply).
Software
- Laptop running MinKNOW software v 0.48.2.12 (ONT) with internet connection (e.g., via 3G wireless router); for current IT requirements please check the ONT internet site (https://nanoporetech.com/community/faqs).
Procedure
- Reverse transcription (RT) using the SuperScript III First-Strand-Synthesis System
- Combine 4 μl RNase free water, 1 μl primer (2 μM, CGGACACACAAAAAGAAAGAAG), 1 μl dNTPs, 5 μl RNA, mix by flicking, spin down, incubate for 5 min at 65 °C, put in PCR cooler for > 1 min.
- Prepare mastermix (6 μl 5x FS buffer, 1.5 μl DTT, 1.5 μl RNase out, 1.5 μl Superscript III. All components are provided as part of the RT kit), add 7 μl to samples, mix by flicking, spin down, incubate for 50 min at 50 °C , 5 min at 85 °C, hold at 4 °C, add 1 μl RNase H, mix by flicking, spin down, incubate for 20 min at 37 °C, hold at 4 °C.
- Combine 4 μl RNase free water, 1 μl primer (2 μM, CGGACACACAAAAAGAAAGAAG), 1 μl dNTPs, 5 μl RNA, mix by flicking, spin down, incubate for 5 min at 65 °C, put in PCR cooler for > 1 min.
- PCR using iProof polymerase
- Set up mastermix for 14 reactions (Table 2; this includes 2 extra reactions that are not used) using the cDNA from step A2 as template, dispense 12 x 44.2 μl into 0.2 ml PCR tube strips on PCR cooler block, add 2.5 μl forward primer (10 μM) and 2.5 μl reverse primer (10 μM) according to table 1, flick, spin down, cycle (Table 3, first PCRs).
- Set up mastermix for 28 reactions (Table 2; this includes 4 extra reactions that are not used) using the PCR products from step B1 as template, dispense 24 x 44.2 μl into PCR tube strips on PCR cooler block, add 0.8 μl PCR product from step B1 as template (see Table 1 to determine which PCR product from step B1 to use as template), add 2.5 μl forward primer (10 μM) and 2.5 μl reverse primer (10 μM) according to Table 1, flick, spin down, cycle (Table 3, second PCRs). For an overview of the whole procedure see Figure 1.
Table 1. Primer sequences. Each pair of primer sequences (1 forward and 1 reverse primer) correspond to a single reaction.
Table 2. PCR master mix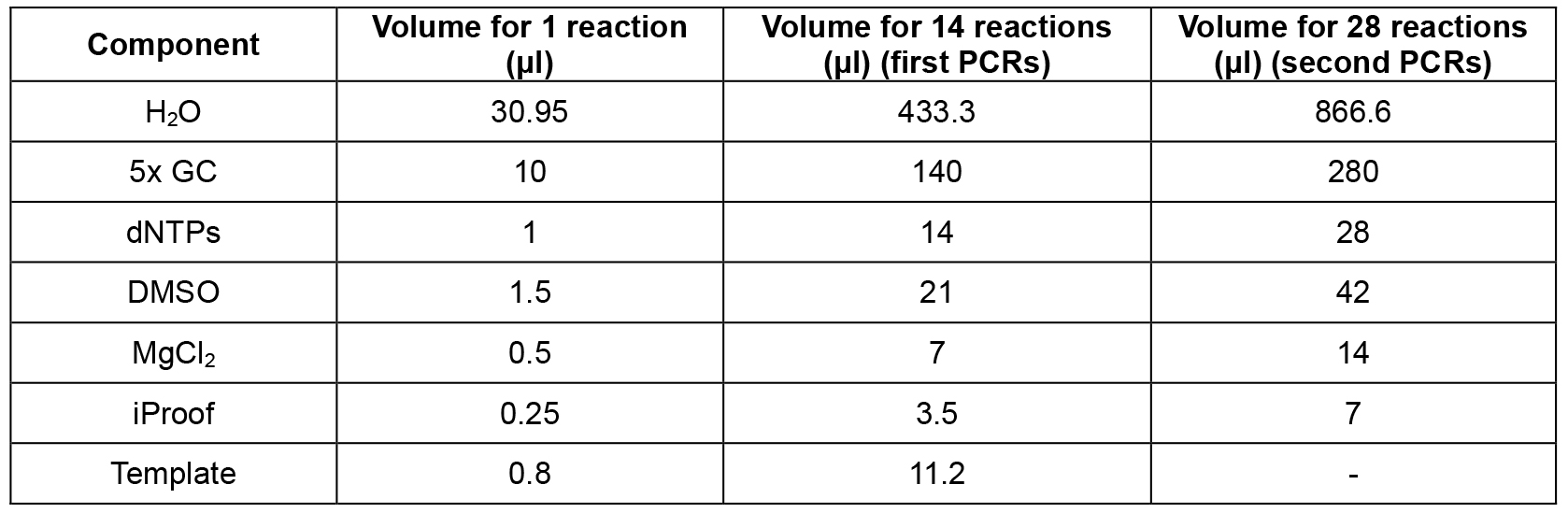
Table 3. PCR cycling conditions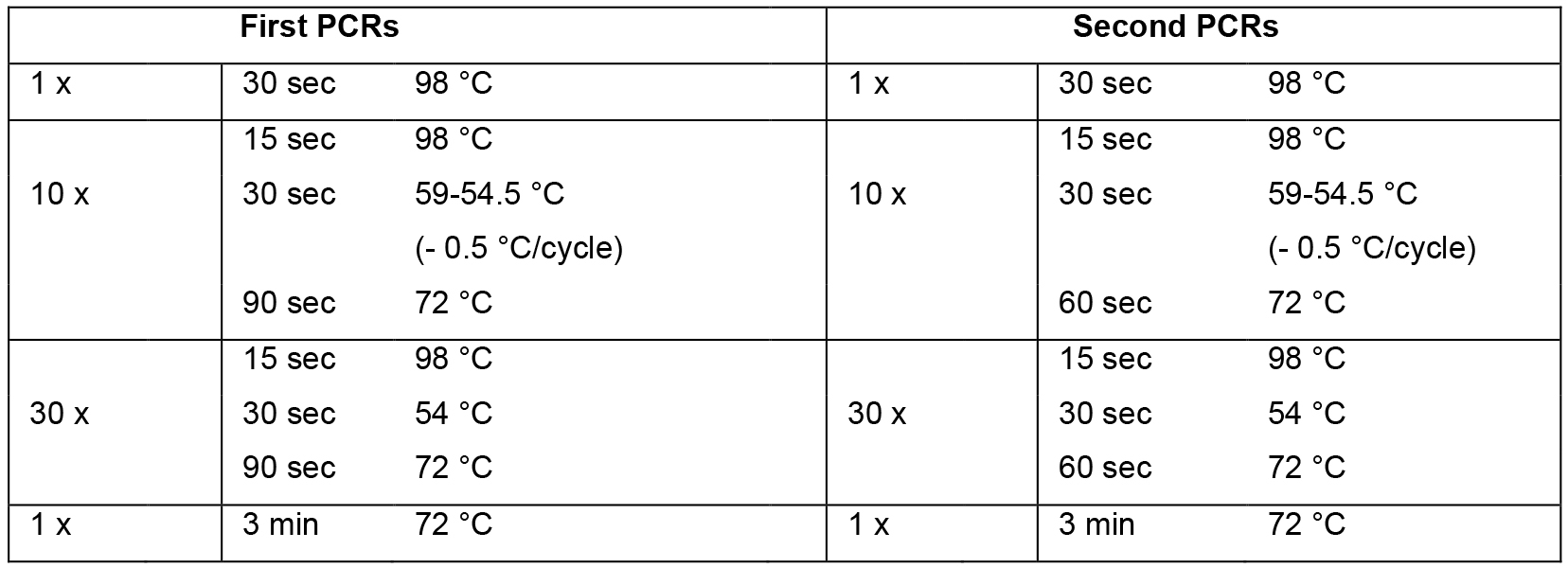
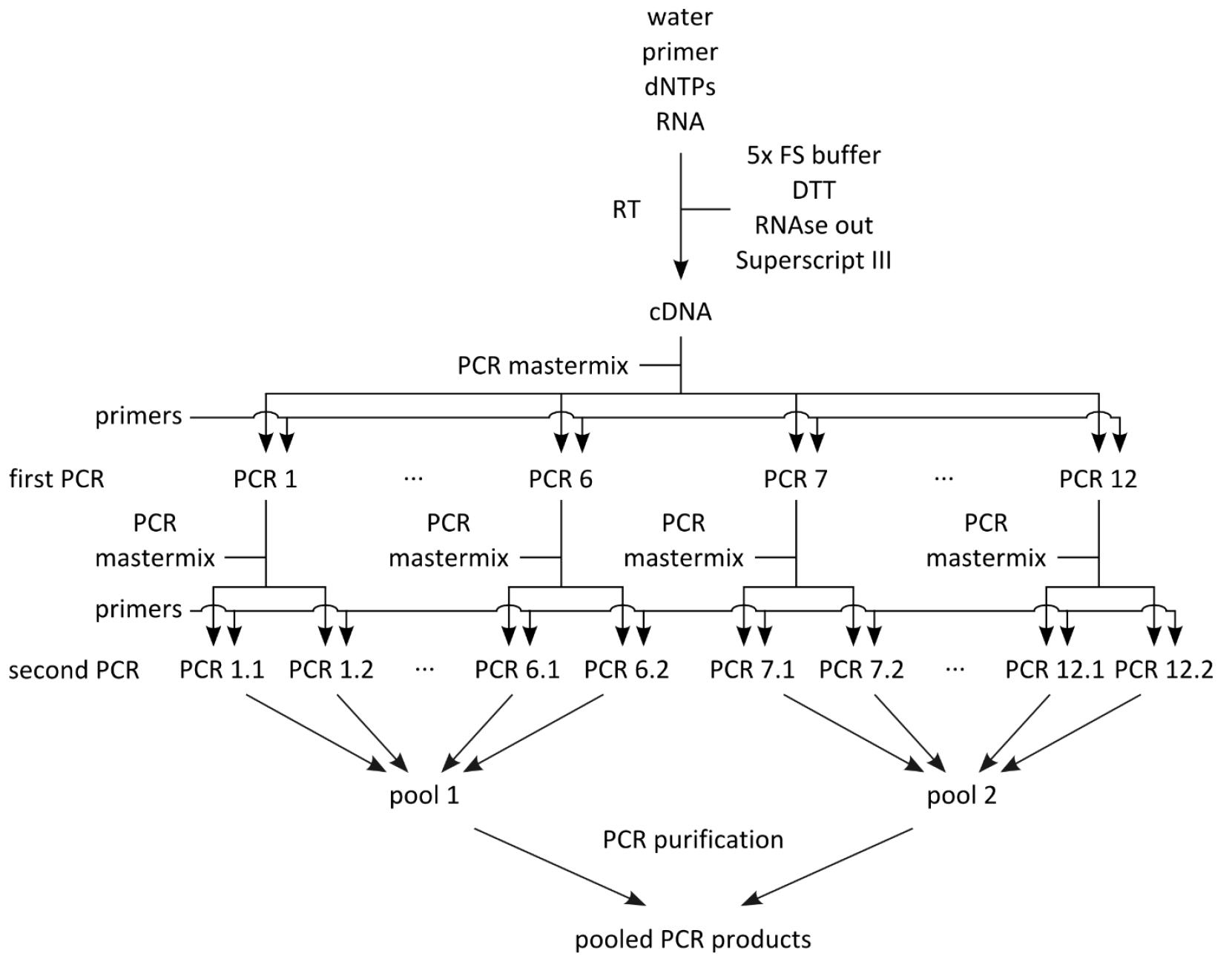
Figure 1. Schematic of PCR amplification prior to library preparation
- Set up mastermix for 14 reactions (Table 2; this includes 2 extra reactions that are not used) using the cDNA from step A2 as template, dispense 12 x 44.2 μl into 0.2 ml PCR tube strips on PCR cooler block, add 2.5 μl forward primer (10 μM) and 2.5 μl reverse primer (10 μM) according to table 1, flick, spin down, cycle (Table 3, first PCRs).
- Agencourt PCR-purification
- Vortex AMPure XP beads for 10 sec.
- Out of the 24 PCR products, generate two pools of PCR products (Figure 1; the first pool contains PCR products 1.1-6.2, and the second pool PCR products 7.1-12.2), each containing 12 x 40 μl, for a total of 480 μl each, and pipet into 1.5 ml Eppendorf tube.
- Add 720 μl beads to the pools, mix by pipetting 10 times, incubate for 5 min.
- Place tube onto magnet, wait 5 min.
- Take off supernatant completely, being careful not to aspirate any beads, add 1,200 μl 70% EtOH (do NOT pipet directly onto beads), let sit for at least 30 sec, take off completely with a P1000 pipette.
- Repeat wash step with 800 μl 70% EtOH, take off supernatant with a P1000 pipette, spin down for 3 sec, put back on magnet, take off any remaining supernatant with a P10 pipette.
- Dry beads for about 2 min with open lids, keep lids open from now on.
- Take tube off magnet, add 60 μl Qiagen elution buffer, resuspend bead-pellet in elution buffer, incubate for 10 min at room temperature.
- Place back onto magnet, wait 1 min (until beads have pelleted), take off 50 μl of eluate. Pool the eluates from the two purified PCR product pools, for a total of 100 μl containing all PCR products.
- Vortex AMPure XP beads for 10 sec.
- Optional: Quality control on agarose gel
- Prepare 1x TAE by adding 10.2 ml 50x TAE into 500 ml water.
- Combine 50 ml 1x TAE and 0.5 g agarose, bring to boil in microwave.
- Pour agarose gel, let solidify on a cold pack.
- Load 15 μl of DNA ladder and 5 μl of pooled sample mixed with 1 μl 6x loading dye onto agarose gel, run for about 20 min at 100 V.
- Combine 50 ml FastBlast DNA stain and 200 ml water, incubate the agarose gel for 3 min in the diluted DNA stain, then rinse and destain the gel with several changes of water, rocking the gel continuously for 10 to 20 min.
- Visualize the gel using a white laptop screen as lightbox. Several discrete bands should be visible, corresponding to the various PCR product lengths (strong band at ~2.1 kb, moderate band at ~2.7 kb, weak bands at 1.3, 3.4, and 4.0 kb).
- Prepare 1x TAE by adding 10.2 ml 50x TAE into 500 ml water.
- Library preparation
- Combine 80 μl of pooled, purified PCR products from step C9 and 5 μl DNA CS, add 10 μl 10x NEBNext end repair buffer and 5 μl NEBNext end repair enzyme mix, mix by pipetting, incubate at RT for 30 min.
- Clean up with 180 μl Agencourt beads as described in section C, but use 300 μl EtOH for washing, add 30 μl elution buffer, take off 25 μl eluate, put in PCR tube.
- Add 3 μl 10x NEBNext dA-tailing buffer and 2 μl NEBNext dA-tailing enzyme mix, incubate for 30 min at 37 °C.
- Thaw 2x wash buffer, elution buffer, EP buffer at RT, thaw HP adapter, Adapter mix and Fuel mix in the cooler box.
- Prepare His-beads:
- Mix 550 μl 2x wash buffer and 550 μl nuclease-free water by inverting 10x, briefly spin down.
- Vortex His-Beads for ~30 sec.
- Pipet 10 μl His-Beads into a LoBind tube, add 250 μl 1x wash buffer.
- Place on magnet, wait for pellet to form, aspirate off supernatant, add 250 μl wash buffer, wait 30 sec, aspirate off supernatant.
- Take tube off magnet, resuspend pellet in 100 μl 2x wash buffer.
- After 30 min, in a LoBind tube, add (directly to the bottom of the tube) 8 μl nuclease-free water, 30 μl DNA from step E3, 10 μl Adapter mix, 2 μl HP Adapter, and 50 μl Blunt/TA ligase master mix, mix by pipetting, if necessary briefly spin down, incubate 10 min at RT.
- Add 100 μl washed beads (directly to bottom of tube), carefully mix by pipetting, incubate for 5 min at RT.
- Place on magnet, wait 2 min for pellet to form, aspirate supernatant, wash 2 x for 30 sec each with 250 μl 1x wash buffer.
- With the lid close, briefly centrifuge, put back onto magnet, wait 1 min with lid closed, aspirate off any remaining wash buffer.
- Take off magnet, resuspend pellet in 25 μl elution buffer (adding the elution buffer directly to the pellet), wait 10 min with lid closed, put back on magnet, wait 1 min, take off 15 μl eluate (≥ pre-sequencing mix).
- Combine 80 μl of pooled, purified PCR products from step C9 and 5 μl DNA CS, add 10 μl 10x NEBNext end repair buffer and 5 μl NEBNext end repair enzyme mix, mix by pipetting, incubate at RT for 30 min.
- Loading of library (Figure 2)
- Mix 318.5 μl EP buffer and 6.5 μl Fuel mix by vortexing, spin down.
- Prime flow cell two times with 150 μl of EB/Fuel mix.
- Mix 6 μl library, 141 μl EP, 3 μl Fuel mix by inverting 10 times, briefly spin down, load, and initiate flow cell run.

Figure 2. Priming/loading of the flowcell
- Mix 318.5 μl EP buffer and 6.5 μl Fuel mix by vortexing, spin down.
Data analysis
Once the sequencing run is completed on the MinION device, raw data have to be base-called, primer sequences have to be removed, and then all sequences have to be aligned to a reference sequence to build a pile-up, which can then be used for consensus-calling. The exact procedure for doing so, including all used bioinformatics scripts, is published in (Hoenen et al., 2016), which is openly accessible at this link: http://wwwnc.cdc.gov/eid/article/22/2/15-1796_article.
Acknowledgments
The author is grateful to all members of the former NIH/CDC diagnostic laboratory at ELWA3, Monrovia, Liberia. This work was funded in part by the Intramural Research Program of the National Institutes of Health, NIAID. The author was participant of the MinION access program, and received some of the flowcells and reagents used from ONT free of charge or at reduced cost. The author was invited by ONT to present part of this work at the ‘London Calling’ 2015 meeting organized by ONT in London, U.K.
References
- de Wit, E., Falzarano, D., Onyango, C., Rosenke, K., Marzi, A., Ochieng, M., Juma, B., Fischer, R. J., Prescott, J. B., Safronetz, D., Omballa, V., Owuor, C., Hoenen, T., Groseth, A., van Doremalen, N., Zemtsova, G., Self, J., Bushmaker, T., McNally, K., Rowe, T., Emery, S. L., Feldmann, F., Williamson, B., Nyenswah, T. G., Grolla, A., Strong, J. E., Kobinger, G., Stroeher, U., Rayfield, M., Bolay, F. K., Zoon, K. C., Stassijns, J., Tampellini, L., de Smet, M., Nichol, S. T., Fields, B., Sprecher, A., Feldmann, H., Massaquoi, M. and Munster, V. J. (2016). The merits of malaria diagnostics during an Ebola virus disease outbreak. Emerg Infect Dis 22(2): 323-326.
- Hoenen, T., Groseth, A., Rosenke, K., Fischer, R. J., Hoenen, A., Judson, S. D., Martellaro, C., Falzarano, D., Marzi, A., Squires, R. B., Wollenberg, K. R., de Wit, E., Prescott, J., Safronetz, D., van Doremalen, N., Bushmaker, T., Feldmann, F., McNally, K., Bolay, F. K., Fields, B., Sealy, T., Rayfield, M., Nichol, S. T., Zoon, K. C., Massaquoi, M., Munster, V. J. and Feldmann, H. (2016). Nanopore sequencing as a rapidly deployable Ebola outbreak tool. Emerg Infect Dis 22(2): 331-334.
- Quick, J., Loman, N. J., Duraffour, S., Simpson, J. T., Severi, E., Cowley, L., Bore, J. A., Koundouno, R., Dudas, G., Mikhail, A., Ouedraogo, N., Afrough, B., Bah, A., Baum, J. H., Becker-Ziaja, B., Boettcher, J. P., Cabeza-Cabrerizo, M., Camino-Sanchez, A., Carter, L. L., Doerrbecker, J., Enkirch, T., Garcia-Dorival, I., Hetzelt, N., Hinzmann, J., Holm, T., Kafetzopoulou, L. E., Koropogui, M., Kosgey, A., Kuisma, E., Logue, C. H., Mazzarelli, A., Meisel, S., Mertens, M., Michel, J., Ngabo, D., Nitzsche, K., Pallasch, E., Patrono, L. V., Portmann, J., Repits, J. G., Rickett, N. Y., Sachse, A., Singethan, K., Vitoriano, I., Yemanaberhan, R. L., Zekeng, E. G., Racine, T., Bello, A., Sall, A. A., Faye, O., Faye, O., Magassouba, N., Williams, C. V., Amburgey, V., Winona, L., Davis, E., Gerlach, J., Washington, F., Monteil, V., Jourdain, M., Bererd, M., Camara, A., Somlare, H., Camara, A., Gerard, M., Bado, G., Baillet, B., Delaune, D., Nebie, K. Y., Diarra, A., Savane, Y., Pallawo, R. B., Gutierrez, G. J., Milhano, N., Roger, I., Williams, C. J., Yattara, F., Lewandowski, K., Taylor, J., Rachwal, P., Turner, D. J., Pollakis, G., Hiscox, J. A., Matthews, D. A., O'Shea, M. K., Johnston, A. M., Wilson, D., Hutley, E., Smit, E., Di Caro, A., Wolfel, R., Stoecker, K., Fleischmann, E., Gabriel, M., Weller, S. A., Koivogui, L., Diallo, B., Keita, S., Rambaut, A., Formenty, P., Gunther, S. and Carroll, M. W. (2016). Real-time, portable genome sequencing for Ebola surveillance. Nature 530(7589): 228-232.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hoenen, T. (2016). Sequencing of Ebola Virus Genomes Using Nanopore Technology. Bio-protocol 6(21): e1998. DOI: 10.21769/BioProtoc.1998.
Category
Microbiology > Microbial genetics > RNA
Molecular Biology > RNA > RNA sequencing
Systems Biology > Genomics > Sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.