- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Primary Explosive Blast-induced Traumatic Brain Injury Model in PC12 Cell Culture



Published: Vol 6, Iss 16, Aug 20, 2016 DOI: 10.21769/BioProtoc.1907 Views: 7564
Reviewed by: Soyun KimPengpeng LiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
While it is understood that structural damage occurs at the cellular level from the traumatic brain injury event, the effect on functional activity remains largely unknown. Simplified models such as in vitro models of primary explosive blast are critically needed to deconvolute mechanisms of cellular damage. This protocol details an in vitro indoor experimental system setup (Zander et al., 2015) using real military explosive charges to more accurately represent battlefield blast exposure, and probe the effects of primary explosive blast on dissociated neurons.
Keywords: Primary explosive blast modelMaterials and Reagents
- 12 mm circular glass coverslips (Fisher Scientific, catalog number: 12-545-80 )
- 24-well plates (Corning, FalconTM, catalog number: 351147 )
- Sterile SealPlate® covers (Excel Scientific)
- PC12 cells (ATCC, catalog number: CRL-1721 )
- Mouse laminin (Corning, catalog number: 354239 )
- 100% ethanol
- Polylysine (Sigma-Aldrich, catalog number: P5899 )
- RPMI 1640 (Corning, catalog number: 10-041-CV )
- Horse serum (Corning, catalog number: 35-030-CV )
- Fetal bovine serum (FBS) (Fisher Scientific, catalog number: SH3007003IH )
- Dulbecco Modified Eagle’s Medium (DMEM) (Corning, catalog number: 10-013-CV )
- Calf serum (GE Healthcare Life Sciences, HycloneTM, catalog number: SH30072.03 )
- Nerve growth factor (NGF) (Corning, catalog number: 356004 )
- Spherical 1.77 grams/cm3 cyclotrimethylene trinitramine Class 5 (RDX Class V) charges
- HEPES (Fisher Scientific, catalog number: BP299500 )
- 10-gallon poly (methyl methacrylate) (PMMA)
- Calcein-AM (Thermo Fisher Scientific, catalog number: C3099 )
- Ethidium homodimer-1 (Thermo Fisher Scientific, catalog number: E1169 )
- Phosphate buffered saline (PBS) (Fisher Scientific, catalog number: BP399500 )
- Lactate dehydrogenase cytotoxicity assay kit (LDH) (Abnova, catalog number: KA0785 )
- RIPA buffer (Alfa Aesar, catalog number: J62189-AP )
- Protease inhibitor cocktail (Sigma-Aldrich, catalog number: P8340 )
- The micro BCA protein assay kit (Thermo Fisher Scientific, catalog number: 23235 )
- Hank’s Balanced Salt Solution (HBSS) (Corning, catalog number: 21-022-CV )
- Calcein (MP Biomedicals, catalog number: 190167 )
- Penicillin/Streptomycin, 50x (Corning, catalog number: 30-001-Cl )
- Antibiotic/antimycotic (see Recipes)
- Growth/complete medium (see Recipes)
- Differentiation medium (see Recipes)
Equipment
- Incubator
- Aquarium
- Three piezoelectric high frequency dynamic pressure sensors (PCB Piezotronics, model: 102A )
- Sterile hood
- Oven
- Zeiss LSM5 Pascal equipped with Epiplan-Neofluar lenses
- Confocal laser scanning microscopy (CLSM)
Software
- ImageJ v 1.34
- Zeiss LSM software (v 4.2.0.121)
Procedure
- Experimental setup and preparation
- Cell sample preparation
- Clean 12 mm circular glass coverslips using piranha etch [H2SO4:H2O2 = 70:30 (v/v)] for 30 min and then rinse thoroughly with deionized (DI) water.
- Sonicate slides 10 min/time, 3 times in 100% ethanol to sterilize.
- Place sterile coverslips in 24-well plates and coat in 100 µg/ml sterile polylysine solution for 30 min.
- Rinse the slides three times in sterile DI water and allow to air dry.
- Incubate the slides in 10 µg/ml sterile laminin at 4 °C overnight.
- After protein attachment, rinse the slides three times with sterile PBS and use immediately for cell culture.
- Culture cells in RPMI 1640 medium supplemented with 10% heat-inactivated horse serum and 5% fetal bovine serum and 1% antibiotic/antimycotic “complete medium” at 37 °C and 5% CO2. Culture cells according to the supplier’s instructions.
- Seed cells at a density of 5,000 cells/well on the 12 mm coverslips in the 24-well plates in high-glucose DMEM with 1% horse serum, 0.5% calf serum and 1% antibiotic/antimycotic “differentiation medium”.
- After 24 h, add 100 ng/ml NGF to the differentiation medium. Subject the cells to blast 5 days after adding NGF.
- Clean 12 mm circular glass coverslips using piranha etch [H2SO4:H2O2 = 70:30 (v/v)] for 30 min and then rinse thoroughly with deionized (DI) water.
- Blast induced injury in vitro setup
- Use a spherical 1.7 g/cm3 cyclotrimethylene trinitramine Class 5 charge to generate the blast (Figure 1).

Figure 1. Explosive charge
- The charge standoff distance to the neuron cell chamber is measured as a clear spacing between the charge and the outer wall of the aquarium.
- Adjust the explosive charge standoff distance to generate different pressure exposure inside the cell culture wells. For multiple blast exposure, keep the samples inside the tank, and separate each exposure by 5-7 min intervals.
Note: The time interval does not necessarily reflect the actual time between blasts on the battlefield. The interval is the minimum amount of time needed to clear the blast chamber for re-entry and set-up the next explosive. - Three piezoelectric high frequency dynamic pressure sensors were used to measure the shockwave overpressure duration above the cultured samples (Figure 2).

Figure 2. Pressure Sensor Holder. A. Piezoelectric pressure sensors in horizontal position inside testing aquarium. B. Piezoelectric pressure sensors in vertical position outside testing aquarium. - All pressure gauges were mounted on top of the cell culture plate with a custom-designed lid, submerged approximately 4 inches underwater, and positioned side-on to the blast wave direction.
- The caps were designed such that the pressures in different rows or columns of the well plate could be measured by moving the pressure sensors to the desired locations.
- Two pencil gauges were positioned in front of the aquarium to measure the pressure in air before the shock wave moves into the water interface (Figures 3 and 4).

Figure 3. Blast induced cell injury overview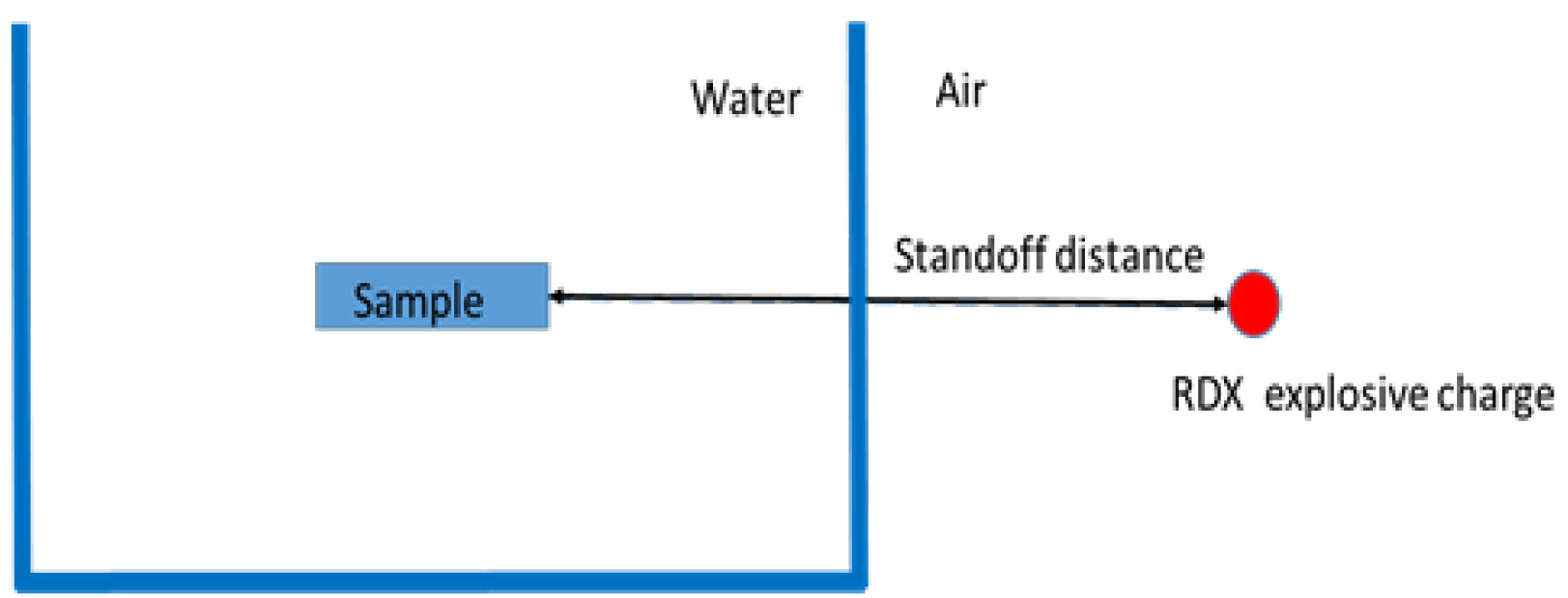
Figure 4. Schematic diagram showing positions of RDX explosive charge, aquarium tank, and sample - Pressure-time history and peak pressure were recorded for each blast.
- Blast-induced injury of cells
- Just prior to the blast experiment in a sterile hood, remove the plastic well plate lid in a sterile hood and add sterile HEPES buffer (10 mM) in the well plate. Seal the well plates with sterile SealPlate® covers. Placed in a plastic bag to maintain sterility of the cultures. Transport the samples to the blast site and put the samples in an oven at 37 °C until the experiments.
- Submerge the samples horizontally on a sample holding stage in a 10-gallon poly (methyl methacrylate) (PMMA) aquarium containing water heated to 37 °C, as displayed in Figure 2.
- Mount, secure, and immerse under water horizontally the neuronal cell line culture well plates in the middle of the aquarium with the cells in the first row (1) facing the shock front.
- All blast experiments are performed without the well plate lid.
- Put the control samples in the incubator for the duration of the experiment.
- Put sham cells in the aquarium for 20 min (typical length of a triple blast experiment).
- Just prior to the blast experiment in a sterile hood, remove the plastic well plate lid in a sterile hood and add sterile HEPES buffer (10 mM) in the well plate. Seal the well plates with sterile SealPlate® covers. Placed in a plastic bag to maintain sterility of the cultures. Transport the samples to the blast site and put the samples in an oven at 37 °C until the experiments.
- Pressure measurements during the blast cell exposure (Figure 5)
Subject the cells to single or multiple blast insults five days after seeding cells in differentiation medium supplemented with 100 ng/ml NGF. To minimize the movement, ensure good attachment of the cells to the coverslip. In addition, the dimensions of the well plate and coverslips need to be chosen carefully so that the coverslip fits tightly within the well.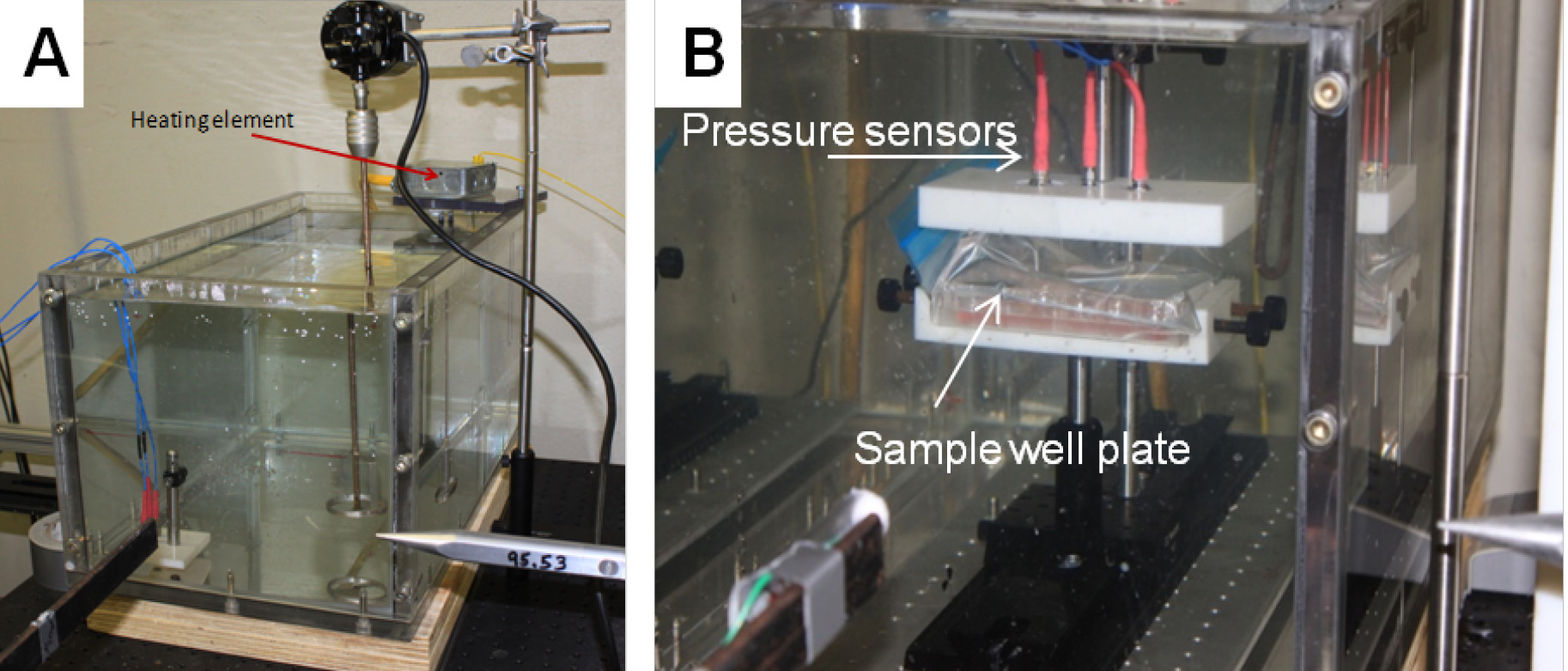
Figure 5. Blast-induced injury of cells. A. Aquarium tank showing pencil gauge. B. Sample well plate submerged in aquarium tank showing location of pressure sensors on lid.
- Cell sample preparation
- Cell analysis
- Cell morphology and viability analysis
For single blast exposure, assess the cell viability at 2 and 24 h post-blast by staining the cells with calcein-AM and ethidium homodimer-1, following the protocol outlined by Life TechnologiesTM.- Briefly, remove the medium and rinse the cells with PBS three times.
- Incubate the cells in a PBS solution containing 2 µM calcein-AM and 4 µM ethidium homodimer-1 for 30 min at 37 °C.
- Using the 543 nm and 488 nm lasers for the viability assay: Image samples by confocal laser scanning microscopy (CLSM) on a Zeiss LSM5 Pascal equipped with Epiplan-Neofluar lenses. Image the cells in the multi-track mode with a 543-nm laser with a minimum of n = 5 random areas for each of a minimum of 3 replicate samples using the 10x and 20x objectives.
- CLSM are processed using image area analysis tools in ImageJ v 1.34, National Institutes of Health. Convert images to binary and thresholds set for each channel (live cells and dead cells).
- Calculate the percent area using the analyze particles tool.
- Correct the smaller cell size for the dead cells compared to live cells by multiplying the percentage of area of dead cells by the average live cell size/average dead cell size.
- Calculate percentage of live cells by taking the live cell percent area over the total live and corrected dead cell percent area.
- Assess neurite morphology via the calcein AM stain (Figure 6).
- Measure the length and bead diameter using the Zeiss LSM software.
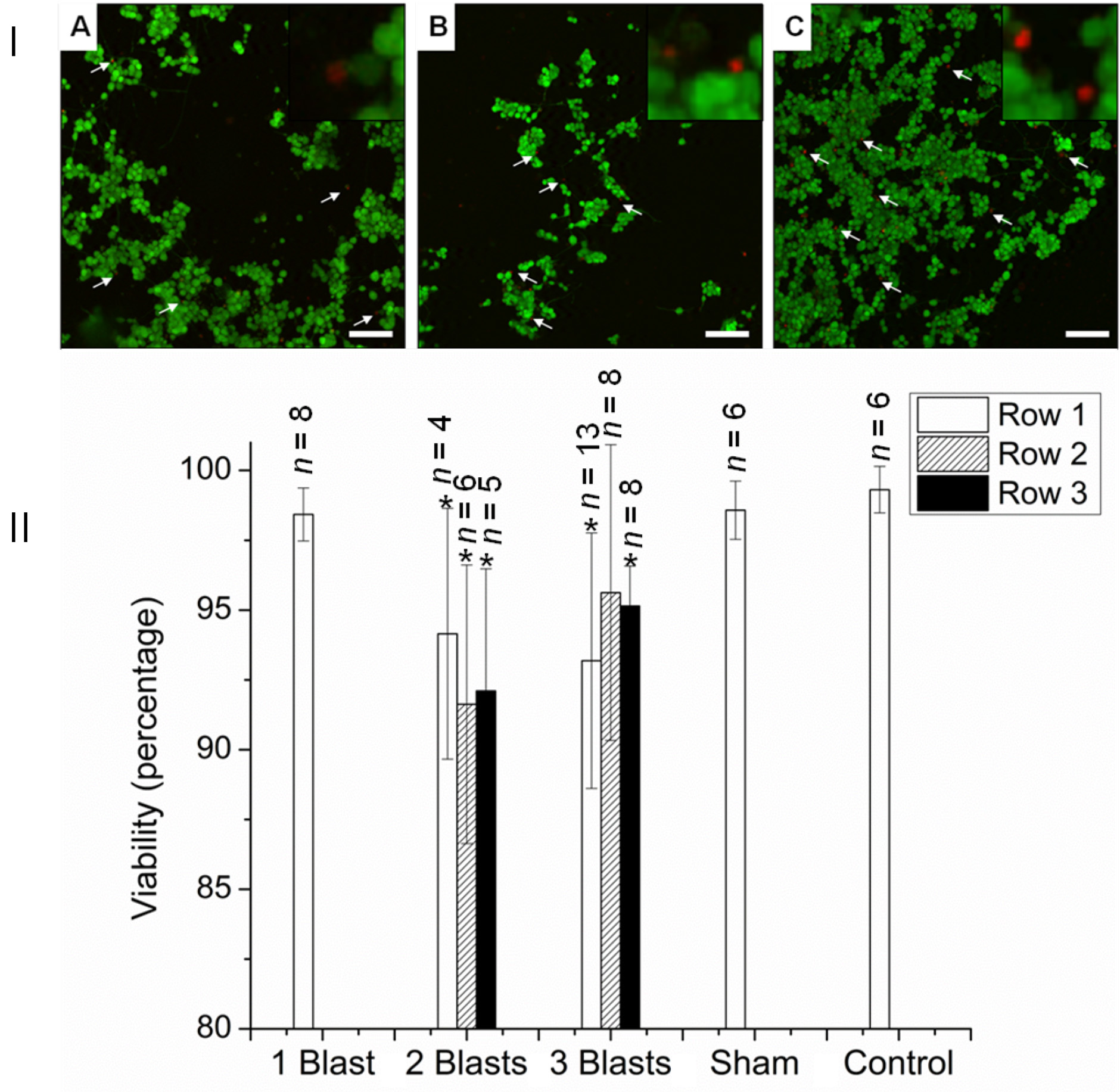
Figure 6. Viability of PC12 neurons 24 h after exposure to single and multiple explosive blasts of ca. 32 psi. I: CSLM images with live/dead stain. (A) One blast, (B) Two blasts, (C) Three blasts. Arrows indicate selected dead cells. Insets: magnified images of dead cells. II. Viability quantification. *P < 0.05 compared with control, sham, and one blast. Scale bars = 100 µm in A-C; 50 µm for insets - Briefly, remove the medium and rinse the cells with PBS three times.
- Membrane permeability assays
- Sample media from the extracellular bath at 1, 2, 4 and 24 h post-blast.
- Perform the LDH assay according to the manufacturer’s instructions.
- Remove the media and rinse the samples with PBS and dry the coverslips for few minutes.
- Add approximately 20 µl of RIPA buffer with 1% (vol) protease inhibitor to each coverslip.
- Scrap the cells immediately and analyze using the Micro BCA protocol or put on ice for 30 min and then stored at -80 °C until analysis.
- Normalize all values to the total protein determined by a micro BCA protein assay.
- Probe calcein uptake by rinsing the samples with HBSS (without calcium and magnesium) and then incubating in 0.3 mM calcien in HBSS for 10 min.
- Rinse the samples thoroughly with HBSS and image using the 488 nm laser on the CLSM system.
- Fix imaging gains and offsets to allow semi-quantitative comparison between samples.
- Collect phase-contrast images to ensure cells are in focus.
- Measure fluorescence intensities by selecting individual cells using the region of interest (ROI) feature and calculating the mean intensity using the histogram feature using the software (Figure 7).
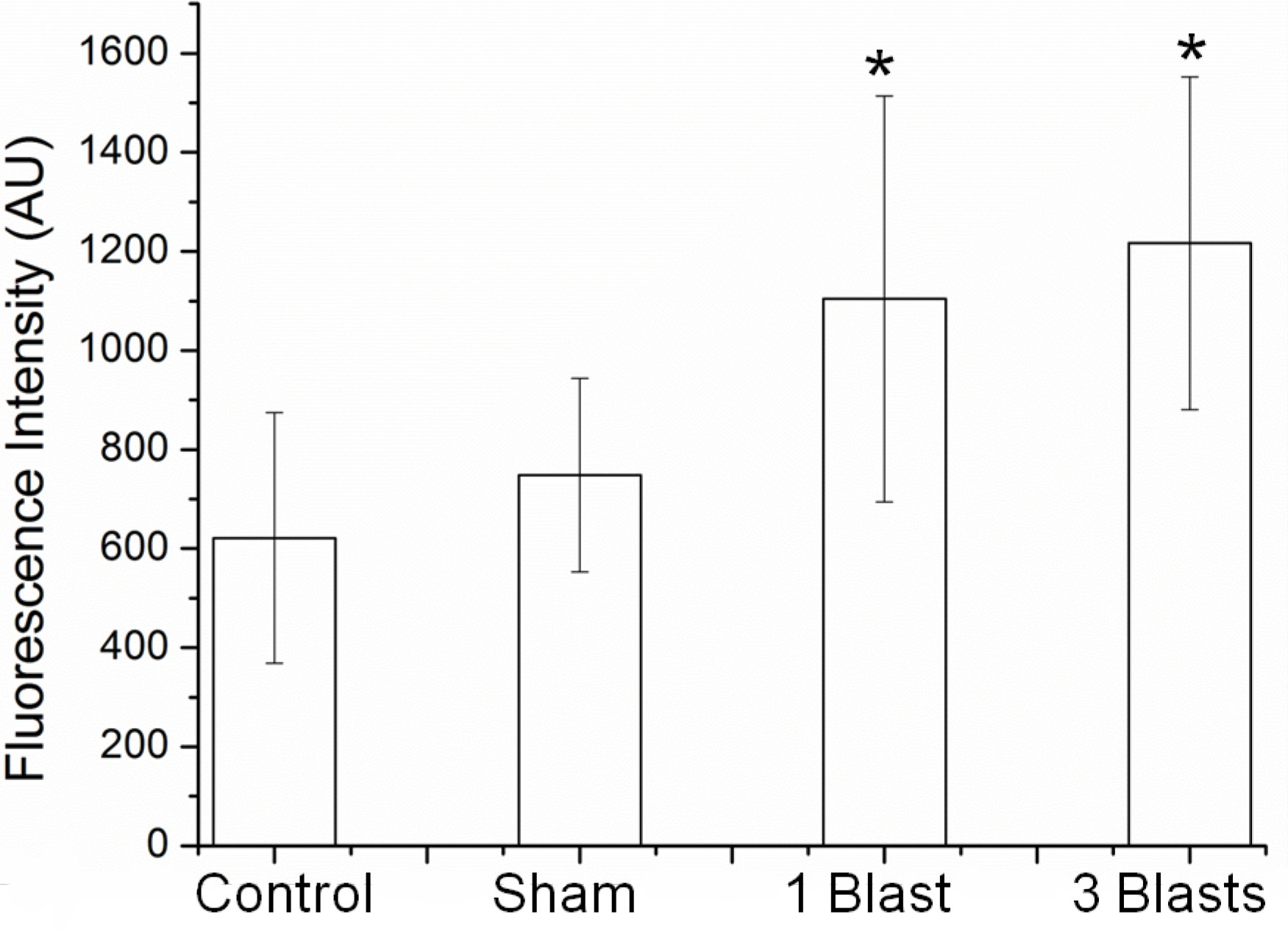
Figure 7. Membrane permeability changes as a function of calcein dye uptake of PC12 neurons exposed to explosive blast of ca. 32 psi. *P < 0.05 compared with control and sham, n = 3 - Sample media from the extracellular bath at 1, 2, 4 and 24 h post-blast.
- Cell morphology and viability analysis
Recipes
- Antibiotic/antimycotic
10,000 IU penicillin
10,000 µg/ml streptomycin
25 g/ml amphotericin - Growth/complete medium
High-glucose DMEM
10% heat-inactivated horse serum
5% fetal bovine serum
1% antibiotic/antimycotic - Differentiation medium
High-glucose DMEM
1% horse serum
0.5% calf serum
1% antibiotic/antimycotic
100 ng/ml NGF
Acknowledgments
This research was supported by the U.S. Army Research Laboratory’s Director’s Science Initiative, DSI-13-WMR-2D-005.
References
- Zander, N. E., Piehler, T., Boggs, M. E., Banton, R. and Benjamin, R. (2015). In vitro studies of primary explosive blast loading on neurons. J Neurosci Res 93(9): 1353-1363.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Piehler, T., Zander, N. and Benjamin, R. (2016). Primary Explosive Blast-induced Traumatic Brain Injury Model in PC12 Cell Culture. Bio-protocol 6(16): e1907. DOI: 10.21769/BioProtoc.1907.
Category
Neuroscience > Behavioral neuroscience > Cognition
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.