- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Phagocytosis Assay of Microglia for Dead Neurons in Primary Rat Brain Cell Cultures

Published: Vol 6, Iss 8, Apr 20, 2016 DOI: 10.21769/BioProtoc.1795 Views: 11778
Reviewed by: Oneil G. BhalalaAlka MehraVivien Jane Coulson-Thomas
Original research article
The authors used this protocol in:

Aug 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Clearance of dead brain tissue including the dead neurons through phagocytosis is an endogenous function of microglia in the brain, which is critical for inflammation resolution after ischemic stroke or head trauma. By regulating the function or polarization status of microglia, we may control their phagocytosis efficacy and therefore the cleanup process for the dead brain tissue. We cultured rat cortical neurons and microglia from the same litter of embryos. The cultured neurons are subjected to irradiation for inducing neuronal apoptosis. After labeling with propidium iodide (PI), the dead neurons (DNs) are exposed to the cultured microglia for phagocytosis assay. By counting the number of DNs in each microglia, we calculate the phagocytosis index to quantify the phagocytosis efficacy of microglia toward DNs. The protocol is divided into 4 sections: A) culturing rat cortical neurons from pre-natal rat embryos, B) preparing dead neurons as phagocytosis target, C) culturing rat brain microglia, D) quantifying phagocytosis index of microglia toward the dead neurons.
Keywords: MicrogliaMaterials and Reagents
- Cell Strainer (100 μm and 40 μm) (BD, Falcon, catalog number: 431752 and 431750)
Note: Currently, it is “Corning, Falcon, catalog number: 431752 and 431750 ”. - Cell Lifter (VWR International, catalog number: 89030-910 )
- 100 mm Petri dish (Thermo Fisher Scientific, catalog number: 263991 )
- 60 mm TC dish (Corning, catalog number: CLS430166 )
- 75 cm2 TC flask (Corning, catalog number: 430825 )
- 24-well TC plate (BD, Falcon, catalog number: 353047)
Note: Currently, it is “Corning, Falcon, catalog number: 353047 ”. - 1 ml Syringe (BD, Falcon, catalog number: 305217 )
- Glass Pasteur pipet (VWR International, catalog number: 14673-043 )
- E-18 Pregnant Sprague Dawley (SD) rat (Charles River Laboratories International)
- Pentobarbital (Sigma-Aldrich, catalog number: P-3761 )
- Neurobasal medium (Thermo Fisher Scientific, GibcoTM, catalog number: 21103 )
- B27 Supplement (Thermo Fisher Scientific, GibcoTM, catalog number: 17504 )
- Glutamine (Sigma-Aldrich, catalog number: G7513 )
- Penicillin/Streptomycin (100x) (GE Healthcare, HycloneTM, catalog number: SV30079 )
- DMEM (Corning, catalog number: 10-013-CM )
- Propidium Iodide (PI) (Sigma-Aldrich, catalog number: P-4170 )
- DAPI (4’, 6-Diamidino-2-Phenylindole, Dilactate) (Invitrogen, catalog number: D3571 )
Note: Currently, it is “Thermo Fisher Scientific, Molecular ProbesTM, catalog number: D3571”. - 16% Paraformaldehyde (PFA) (Electron Microscopy Sciences, catalog number: 15170 )
- Fetal Bovine Serum (FBS) (GE Healthcare, HycloneTM, catalog number: SH30071 )
- Sodium pyruvate (Sigma-Aldrich, catalog number: P5280 )
- HEPES Buffer solution (Sigma-Aldrich, catalog number: 83264 )
- Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761 )
- Poly-D-Lysine (Sigma-Aldrich, catalog number: P0889 )
- HBSS (Lonza, catalog number: 10-543F )
- PBS (Corning, catalog number: 21-040-CV )
- 0.4% Trypan blue (Sigma-Aldrich, catalog number: T8154 )
- Alexa Fluor 488-Phalloidin (Invitrogen, catalog number: A12379 )
Note: Currently, it is “Thermo Fisher Scientific, Molecular ProbesTM, catalog number: A12379”. - Mouse anti-MAP2 antibody (Sigma-Aldrich, catalog number: M4403 )
- Rabbit anti-mouse IgG-Alexa Fluor 488 (Thermo Fisher Scientific, InvitrogenTM, catalog number: A11029 )
- Dissection buffer (see Recipes)
- DMEM/FBS (see Recipes)
- Neurobasal/B27 (see Recipes)
Notes:- All reagents and chemicals for cell cultures must be sterile. Without special indication, all culture medium or buffer is used at room temperature (RT). And all operational procedures for neuronal culture and microglia (MΦ) culture, including animal dissecting, tissue triturating, cell plating or harvesting, and medium changing, have to be performed in a dissecting hood or cell culture hood using sterile tools.
- Poly-D-Lysine coated tissue culture (TC) plate. The TC dish and plate for the primary neuronal cells or purified microglia must be pre-coated with Poly-D-lysine (0.1 mg/ml in H2O) (2 ml for 60 mm2 TC dish, 0.5 ml per well for 24w-TC plate) for 30 min at 37 °C. After washing with sterile water, the TC dishes/plates have to be air-dried in the cell culture hood before use.
- The age of the embryos for cell culture. Although E16 embryos are more suitable for neuronal culture and post-natal 1-2 -day-old pups are more suitable for glia culture, we used the E-18 embryos to prepare the neuron culture and also the glial culture (to isolate MΦ). Thus, the neurons and MΦ are prepared from the same litter of embryos. The neuronal culture and microglia can be prepared from embryos or pups of different litters.
- The purity of neurons in the cortical neuronal culture. The Neurobasal/B27 medium is serum-free medium, which is optional for neuronal culture. However, there are other cell types in the cultures. In the 2-day-old neuronal cultures, the ratio of MAP2+-neurons is 43.6 ± 23% (n = 10, Figure 1).
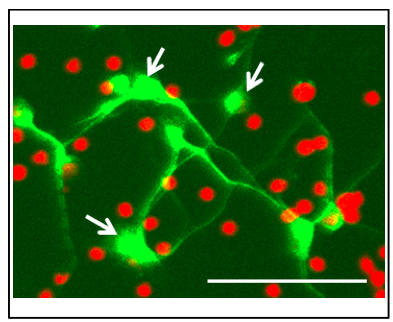
Figure 1. MAP2 immunofluorescence of 2-day-old rat cortical neurons in culture. The cortical neuron culture is fixed in 2% PFA and labeled by mouse anti-MAP2 antibody. The signals are visualized with Alexa Fluor 488 (Green). The nuclei of all cells are labeled with DAPI (red). The arrows indicate the MAP2+-neurons. Scale bar = 50 μm. - All animal studies followed the guidelines outlined in Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and were approved by the Animal Welfare Committee of The University of Texas Health Science Center at Houston.
- The Irradiation operation followed the guidelines for Environment Health & Safety of The University of Texas Health Science Center at Houston.
Equipment
- Hemacytometer (VWR International, catalog number: 15170)
- Cell culture CO2 incubator (LabX, Sanyo, model: MCO-19AIC )
- Fluorescence microscopy with Imaging System, Olympus IX81 controlled by MetaMorph 7.4
- Dissecting microscope (Nikon Corporation, model: SMZ-27 )
- Digital controlled orbital shaker (Thermo Fisher Scientific, model: MaxQTM 2000 )
- 137Cs Irradiation Source (J. L. SHEPHERD & ASSOCIATES, model: 143 ) (Figure 2)

Figure 2. Photos of the 137Cs irradiator. The cultured neurons grown in the 60 mm TC dishes are loaded into the metal cylinder at top of the irradiator. Upon pressing the red button on the right controller, the cylinder will rotate and go down until completely being imbedded in the irradiator (the round bottom portion) for irradiation. By the irradiation time (the left controller) is over, the irradiator will automatically rotate the cylinder upward to its original position. By now, the irradiated cultures are ready to be removed from the cylinder and returned to the CO2 incubator.
Software
- MetaMorph software, version 7.4
Procedure
- Culturing rat cortical neurons
- Deeply anesthetize one E-18 pregnant rat by intraperitoneal injection of sodium pentobarbital (50 mg/kg).
- Prepare the surgical site with an appropriate skin disinfectant. We suggest 70% alcohol.
- Remove the embryos from uterus to one 100 mm Petri dish with 10 ml ice-cold Dissection Buffer.
- Remove the embryos’ brain using two surgical forceps and transfer the brains to another Petri dish with 10 ml ice-cold Dissection Buffer and leave the cells in dish on ice.
- Under a dissecting microscope, after removing olfactory bulbs and meninges, the cortices are separated from other brain regions, and rinsed in ice-cold Neurobasal/B27 medium once. The dissected brain cortices may be divided into two portions: One portion will be used to culture neurons (see next step in this section) and another portion will be used to culture microglia (see Section C).
- Transfer the cortices in a 100 μm cell strainer placed in a 100 mm petri dish containing 10 ml ice-cold Neurobasal/B27 medium and mince the tissue in the same medium by using a 1 ml-syringe plunger.
- The dissociated cells passing through the 100 μm cell strainer (which is placed in a 100 mm petri dish) are applied onto another 40 μm cell strainer, which is placed on top of a 50 ml centrifuge tube.
- The collected cell suspension is subjected to a centrifugation step at 400 x g, 5 min. The pelleted cells are suspended in Neurobasal/B27 medium (from 5 rat embryos) are counted and seeded in Poly-D-lysine coated 60 mm TC dish (5 ml/dish) at a cell density of 2.5 x 107 cells/ml in Neurobasal/B27 medium (Kim et al., 2004). The yield of the cells is about 2.5 x 108 per brain, which can be seeded in 2 of 60 mm TC dishes.
- Culture the cells in a CO2 incubator (5% CO2, 21% O2) at 37.0 ± 0.5 °C for 2 d.
- After two days, the culture medium is completely replaced with fresh one and it is ready for irradiation treatment.
- Preparing dead neurons (DNs) as phagocytosis target
- The 2-day-old neuronal cells grown in 60 mm TC dishes from the above (step A10) are subjected to 137Cs irradiation, for 15 min (32Gy) (Kim et al., 2004). The culture dishes are automatically rotated during radiation to ensure uniform exposure. The irradiation operation is performed with supervision of the staff in the department of the Environment Health and Safety.
- After irradiation, the cells are returned to the original CO2 incubator and cultured in the same culture medium for another 48 h. This incubation time after irradiation injury allows the injured cells progressively undergo apoptosis. Using Trypan Blue, MAP2 immunofluorescence and DAPI staining, we confirmed that ≥95% neurons at this stage are shrunken and dead (Figure 3). By comparing the cell number in the control dish (Figure 3A) and in the irradiation-treated dish (Figure 3B). There is no significant neuronal loss. The immunofluorescence procedures for MAP2 followed the same method as we reported previously (Zhao et al., 2006).
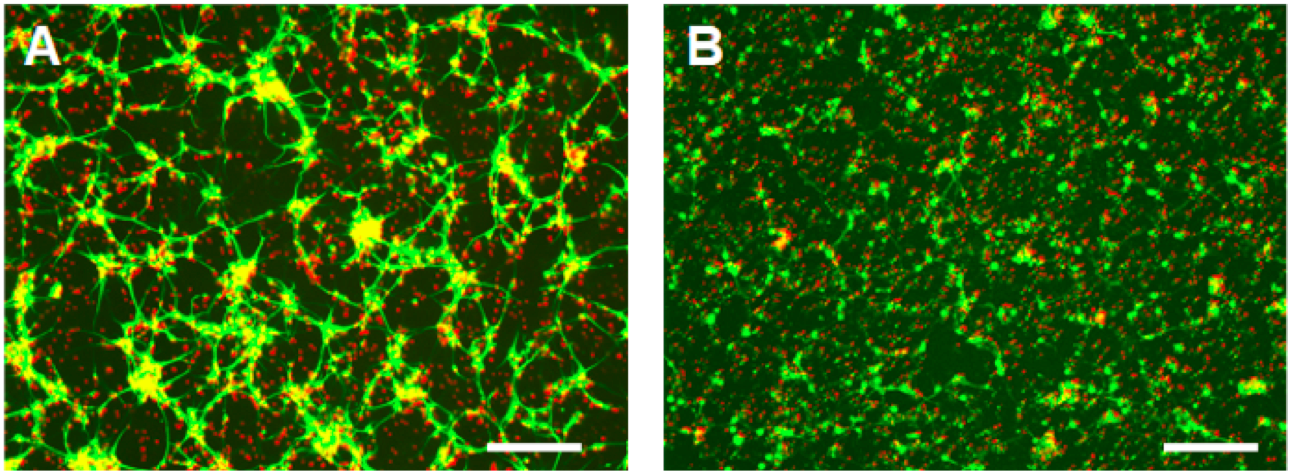
Figure 3. MAP2 immunofluorescence of 4-day-old rat cortical neurons in culture. A. Control; B. 2 d after irradiation injury. The cortical neuron cultures are fixed in 2% PFA and labeled by mouse anti-MAP2 antibody. The signals are visualized with Alexa Fluor 488 (green). The nuclei are highlighted with DAPI (red). Scale bar = 50 μm - Label the dead neurons (DNs) in the culture dishes by adding 1 μg/ml of Propidium Iodide (PI) directly into the culture medium and incubating for 10 min in the CO2 incubator.
- Wash the cells with 10 ml PBS (37 °C), 3 times.
- After removing the PBS, add 3 ml Neurobasal/B27 medium (37 °C) into each dish and harvest the cells by using a Cell Lifter and then collect the cells in a 10 ml centrifuge tube.
- Triturate the cells using a Pasteur Pipet 5-10 strokes to ensure all cells to be dissociated.
- Apply the cell suspension through a 100 μm Cell Strainer and wash the strainer with 2 ml Neurobasal/B27 medium (37 °C).
- Collect all passed cells in one 15 ml centrifuge tube and centrifuge at 400 x g for 10 min.
- Re-suspend the cell pellet in 300 μl of DMEM/FBS per 60 mm TC dish and count the cell number using one hemocytometer. Then, adjust the cell density to 5 x 108 cells/ml in the same culture media. Now the PI-DNs are ready to be used as phagocytosis target at the next step for phagocytosis assay.
- Keep the PI-DNs at 4 °C fridge and wrap the tube with foil to protect the cells from light. The PI-DNs can be safely stored for one month.
- Culturing rat brain microglia (MΦ)
Since we use embryos from the same litter to prepare the cortical neurons (Section A) and also the microglia (Section C), we usually distribute equal number of embryos to each culture. The beginning procedures for preparing microglia cultures are the same as the steps A1-7 described in Section A.- This is next to the step A7 in Section A. The collected cell suspension is subjected to a centrifugation step at 400 x g, 5 min. The pelleted cells are suspended in DMEM/FBS (37 °C) at a ratio of 50 ml per brain and seeded in two 75 cm2 TC flasks (25 ml/flask).
- The glia cells are cultured in CO2 incubator (5% CO2, 21% O2) at 37.0 ± 0.5 °C.
- The culture media are completely changed to fresh one on d 4, d 8 and d 12.
- On day 14, the astrocytes have grown into completely confluent. The MΦ become loosely attached or freely floating in the culture medium. We harvest MΦ by subjecting the flasks to a shaking procedure at 220 rpm for 10 min to detach MΦ from other glia cells.
- Collect the culture medium containing MΦ into 50 ml centrifuge tubes.
- Centrifuge at 400 x g, 10 min.
- After removing the supernatant, re-suspend the pelleted MΦ in 5 ml (per flask) fresh DMEM/FBS (37 °C).
- After cell counting, dilute the cells to 1 x 105 cells/ml with DMEM/FBS. The yield of the MΦ is about 1-2 x 106 cells per flask.
- Re-plate the MΦ in Poly-D-lysine coated 24w-TC plate at 5 x 104 cells in 500 μl/well.
- Culture the purified MΦ in CO2 incubator (5% CO2, 21% O2) at 37.0 ± 0.5 °C for 24 h. And by now it is ready to be used at the next step for phagocytosis assay. By immunofluorescence, we confirmed that more than 95% of the cells are CD68+-MΦ and CD11b+- MΦ (Zhao et al., 2007).
- Quantifying phagocytosis efficacy
- Adding 10 μl of the PI-DNs (prepared at step B10) into the re-plated MΦ cultures (prepared at step C10) at a target/effector ratio of 100:1 (DNs: MΦ).
- Incubate the cells in CO2 incubator (5% CO2, 21% O2) at 37.0 ± 0.5 °C for 2 h.
- At end of incubation, remove the free-floating PI-DNs by aspirating the culture medium and fix the cells with 4% room temperature paraformaldehyde (PFA) for 10 min. It is not necessary to have a washing step prior to fixture.
- Rinse the cells by adding 1 ml PBS in each well, 3 times.
- Incubate the cells in 1 ml of 0.1% Triton X-100 in PBS for 1 min.
- Rinse the cells with 1 ml PBS per well, 3 times.
- Label the cells with 300 μl of Alexa Fluor 488-Phalloidin at 1:100 dilution and 1 μg/ml DAPI in PBS for 30 min.
- Rinse the cells with 1 ml PBS, 3 times.
- Observe the cells under a fluorescence microscopy and acquire the images using the filter sets at Ex/Em of 490/520 nm for Phalloidin-labeled MΦ, Ex/Em of 550/575 nm for PI-DNs and Ex/Em of 365/480 nm for DAPI-labeled nuclei.
- By combining the images acquired with the above 3 sets of filters at the same position, we may clearly observe the PI-DNs within each MΦ. The phagocytized DNs are fluorescently red within Phalloidin-labeled green MΦ. The nuclei of the MΦ are blue (Figure 4). Five images under 40x objective are recorded. The number of the PI-positive neuronal nuclei in each MΦ is counted manually, on still images. At least fifty MΦ cells from each condition should be analyzed. The average number of phagocytized dead neurons (PI-DNs) within each MΦ is calculated and used as Phagocytosis Index (Zhao et al., 2015).
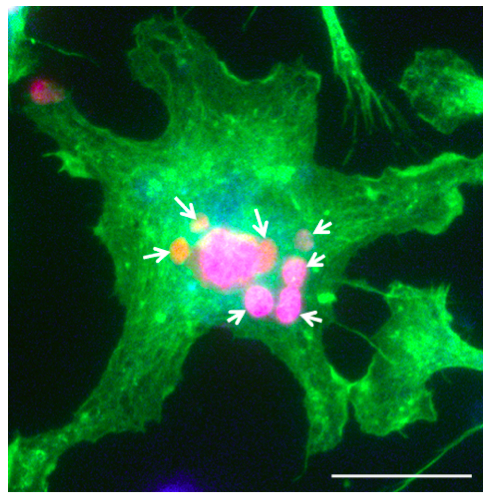
Figure 4. Phalloidin-labeled microglia (green) after phagocytizing the PI-DNs (red). The nucleus is labeled with DAPI (blue). Scale bar = 20 μm. The arrows indicate the phagocytized PI-DNs within the MΦ. - Adding 10 μl of the PI-DNs (prepared at step B10) into the re-plated MΦ cultures (prepared at step C10) at a target/effector ratio of 100:1 (DNs: MΦ).
Notes
- The incubation time of MΦ with dead neurons. We subject MΦ to PI-DNs for 2 h to measure the Phagocytosis Index. To precisely quantify the phagocytosis efficacy, it is necessary to perform a time course (1-4 h) study.
- The number of cell counting. The size of DNs is not unique and the number within each MΦ varies. Therefore, counting the number of DNs in each MΦ should measure all MΦ cells within the image field. We count 50 of MΦ for each experimental condition. However, more number counted, more precise data.
- Bias of image acquisition. To determine the phagocytosis capacity of the MΦ toward the PI-DNs, we suggest using a microscope with a motorized stage and CCD camera, which is controlled by a computer software (e.g., MetaMorph software) that may allow image stitching. This computer-controlled image acquisition system may permit automatic and unbiased cell counting. However, the automatically counted data needs to be confirmed manually. And when multiple batches of cell counting are repeated, it is better done by the same person.
Recipes
- Dissection buffer
HBSS
4.2 mM bicarbonate
1 mM pyruvate
20 mM HEPES
1 mg/ml BSA
1x Penicillin/Streptomycin (pH 7.25) - DMEM/FBS
DMEM
10% FBS
1x Penicillin/Streptomycin - Neurobasal/B27
Neurobasal culture medium
1x B27 supplement
0.5 mM L-Glutamine
1x Penicillin/Streptomycin
Acknowledgments
This protocol was supported in part by grants NS060768, NS064109 and NS084292 of National Institute of Health, National Institute of Neurological Disorders and Stroke. For citation, please refer to Zhao et al. (2015).
References
- Kim, D. H., Zhao, X., Tu, C. H., Casaccia-Bonnefil, P. and Chao, M. V. (2004). Prevention of apoptotic but not necrotic cell death following neuronal injury by neurotrophins signaling through the tyrosine kinase receptor. J Neurosurg 100(1): 79-87.
- Zhao, X., Ou, Z., Grotta, J. C., Waxham, N. and Aronowski, J. (2006). Peroxisome-proliferator-activated receptor-gamma (PPARgamma) activation protects neurons from NMDA excitotoxicity. Brain Res 1073-1074: 460-469.
- Zhao, X., Sun, G., Zhang, J., Strong, R., Song, W., Gonzales, N., Grotta, J. C. and Aronowski, J. (2007). Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol 61(4): 352-362.
- Zhao, X., Wang, H., Sun, G., Zhang, J., Edwards, N. J. and Aronowski, J. (2015). Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci 35(32): 11281-11291.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhao, X., Zhang, L., Ting, S. and Aronowski, J. (2016). Phagocytosis Assay of Microglia for Dead Neurons in Primary Rat Brain Cell Cultures. Bio-protocol 6(8): e1795. DOI: 10.21769/BioProtoc.1795.
- Zhao, X., Wang, H., Sun, G., Zhang, J., Edwards, N. J. and Aronowski, J. (2015). Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci 35(32): 11281-11291.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.