- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Mitochondrial RNA Transcript Analysis Assay of Arabidopsis Leaf Tissues
Published: Vol 5, Iss 20, Oct 20, 2015 DOI: 10.21769/BioProtoc.1620 Views: 9581
Reviewed by: Tie LiuElias BassilAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
This qPCR-based assay provides an overview of the expression levels of all mitochondrial transcripts (mRNAs and rRNAs) as well as splicing efficiency in Arabidopsis. It was developed before RNAseq techniques were widely used (de Longevialle et al., 2007), but is nevertheless still useful as it is cheaper to run and the analysis is much easier and faster to perform if the aim is only to look at mitochondrial transcripts. For intron-containing mRNAs, the use of primer sets specifically amplifying spliced or unspliced forms allows the evaluation of the splicing efficiency.
Keywords: Mitochondrial transcriptomeMaterials and Reagents
- Eppendorf-type microtubes (0.5 ml and 1.5 ml) (any brand)
- LightCycler® 480 Multiwell plate (white) (Roche Diagnostics, catalog number: 04729749001 )
- LightCycler® 480 Sealing Foil for qPCR run (Roche Diagnostics, catalog number: 04729757001 )
- Adhesive PCR film seals for storing plates containing primer mixes (Thermo Fisher Scientific, catalog number: AB0558 )
- Combitips 0.1 ml (Eppendorf, catalog number: 0030 089.405 or 0030 089.618 )
- Arabidopsis thaliana plants grown either in vitro or in soil
- RNeasy Plant Mini Kit (QIAGEN, catalog number: 74904 )
- Water, for molecular biology, DNAse, RNAse and Protease free (Thermo Fisher Scientific, ACROS OrganicsTM, catalog number: 327390010 )
- Ambion Turbo DNA-freeTM Kit (Life Technologies, catalog number: AM1907 )
Note: Currently, it is “Thermo Fisher Scientific, InvitrogenTM, catalog number: AM1907”. - 3M Na acetate (pH 5.2)
- 100 % and 70 % ethanol
- Agarose LE, analytical grade (Promega Corporation, catalog number: V3125 )
- Primers for PCR and qPCR (see Table 1)
Table 1. Primers used in this protocol
Please click here for Table 1. - Taq polymerase
Note: any Taq polymerase can be used for this step, we use a home-purified enzyme. - dNTP set (Life Technologies, catalog number: 10297-018 )
Note: Currently, it is “Thermo Fisher Scientific, InvitrogenTM, catalog number: 10297-018”. - Superscript III Reverse transcriptase (Life Technologies, catalog number: 18080-044 )
Notes: Currently, it is “Thermo Fisher Scientific, InvitrogenTM, catalog number: 18080-044”.
This enzyme comes with 5x transcription buffer and a solution of 0.1 M DTT. - Random Hexanucleotide Primers (Life Technologies, catalog number: 48190011 )
Note: Currently, it is “Thermo Fisher Scientific, InvitrogenTM, catalog number: 48190011”. - RNaseOUT Recombinant Ribonuclease inhibitor (Life Technologies, catalog number:
10777-019 )
Note: Currently, it is “Thermo Fisher Scientific, InvitrogenTM, catalog number: 10777-019”. - LightCycler® 480 SYBR I Master mix (Roche Diagnostics, catalog number: 04887352001 )
Note: This reagent is light sensitive.
Equipment
- Nanodrop UV-Vis Spectrophotometer (Thermo Fisher Scientific)
- ‘Multipette plus’ (Eppendorf, catalog number: 4981 000.019 )
- LightCycler® 480 System (Roche Diagnostics)
Software
- LightCycler® 480 Software version 1.5 (Roche Diagnostics, catalog number: 04994884001)
Procedure
- RNA extraction
- Total Arabidopsis RNA is extracted from young tissue (8 to 20 day-old leaves) with the RNeasy plant mini kit according to the manufacturer’s protocol.
- The RNA is eluted with 40 µl RNase free water and its concentration measured with a NanoDrop.
- Total Arabidopsis RNA is extracted from young tissue (8 to 20 day-old leaves) with the RNeasy plant mini kit according to the manufacturer’s protocol.
- DNase treatment
Genomic DNA needs to be removed prior to the reverse transcription step.
This is achieved using the Ambion Turbo DNA-free kit.- Add in a 0.5 ml tube
A maximum of 10 µg RNA (in 45 µl RNase-free water)
4.7µl 10x Turbo DNase buffer
1 µl DNase - Mix by pipetting and leave tube at 37 °C for 1 h.
- The kit contains a DNase inactivator, which leads to a great loss of material and possible residual resin. Instead, precipitate the reaction with 1/10 vol 3 M Na acetate and 2.5 to 3 volumes of 100% ethanol (30 min to 1h at -20 °C). Spin down at max speed (20,800 x g) for 30 min at 4 °C. Carefully discard the supernatant.
- Wash the pellet with 70% ethanol (carefully add 300 µl of 70% ethanol without disrupting the pellet and spin down for 15 min. This step removes salts, and this will be achieved better if the tube is kept in a -20 °C freezer for 1 h or overnight.
- Dry pellets and resuspend with 30 µl RNase free water.
- Measure concentration with NanoDrop and check RNA quality by running about 100 ng on a 2% agarose-TBE gel. This is a regular DNA gel, just use clean buffer and wash the tray, comb and tank with 0.1% SDS to get rid of RNases. Figure 1A shows intact and degraded leaf RNA samples.
- The absence of contaminating DNA is confirmed by PCR on diluted RNA (1/100) using primers specific for plastid and mitochondrial transcripts.
For example, the pairs Chloro44F and Chloro44R (amplifying rpoB) as well as Mito41F and Mito41R (amplifying cox2) are suitable for this test.The great abundance of plastid DNA in plant cells justifies the use of plastid primers to check the efficiency of the DNase treatment.Chloro44F GCTCCGGAGATAGTTCCCTT rpoB Chloro44R TTCTAGTTCTATCATCAGCTATGGG Mito41F CTCCAGCCGCTCACTGTAAT cox2 Mito41R TCCGATGAGCAGTCACTCAC
Use 1 µl of a 1/100 RNA dilution as template per reaction. Perform the following program: 2 min at 94 °C, 35 cycles (20 sec at 94 °C, 30 sec at 55 °C, 1 min at 72 °C) and finally 5 min at 72 °C.
Do not forget positive (genomic DNA) and negative controls, as ideally this PCR should result in no amplification. When you are satisfied that there is no genomic DNA left in the RNA (see Figure 1B), proceed to cDNA synthesis. Otherwise repeat the DNase treatment.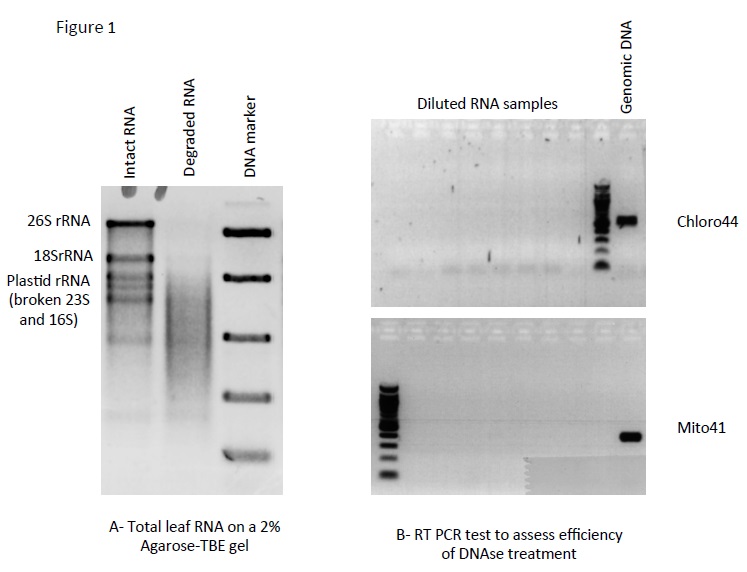
Figure 1. A. TBE gel showing intact (first lane) and degraded (second lane) RNA samples. B. Result of the PCR: the lack of amplification shows that the contaminating DNA has been efficiently removed by the DNase treatment.
- Add in a 0.5 ml tube
- cDNA synthesis
- Use 1-3 µg of DNA-free RNA (0.5 µg is enough in the case of difficult samples), just make sure that the initial quantities of RNA are similar in all samples to be compared and process all samples at the same time.
- It is crucial to use hexanucleotide primers (a mixture of primers comprising six random nucleotides) rather than oligo dT for this step as plastid and plant mitochondrial transcripts do not have polyA tails (unless destined to be degraded). Use the Superscript III First Strand Synthesis System.
- Add in a 0.5 ml tube
0.5 - 3 µg RNA in 11 µl
1 µl 10 mM dNTPs
1 µl random primers at 100 mg/ml
Incubate for 5 min at 65 °C, then incubate on ice for 1-2 min. - Add
4 µl 5x transcription buffer
1 µl 0.1 M DTT
1 µl RNaseOUT
1 µl Superscipt III Reverse Transcriptase
Incubate 5 min at 25 °C
Incubate 50 min at 50 °C
Inactivate the reaction for 15 min at 70 °C and keep the samples on ice or at -20 °C until you are ready to proceed to the qPCR.
- Use 1-3 µg of DNA-free RNA (0.5 µg is enough in the case of difficult samples), just make sure that the initial quantities of RNA are similar in all samples to be compared and process all samples at the same time.
- Quantitative RT-PCR-general considerations
- The cDNAs can be tested by a first round of qPCR using one pair of nuclear primers and one pair of mitochondrial primers from the assay (see Table 1). This will allow you to adjust the cDNA concentrations (by comparing the crossing points (Cp) for all samples) before setting up 3 or 6 x 384-well plates (Figure 2).
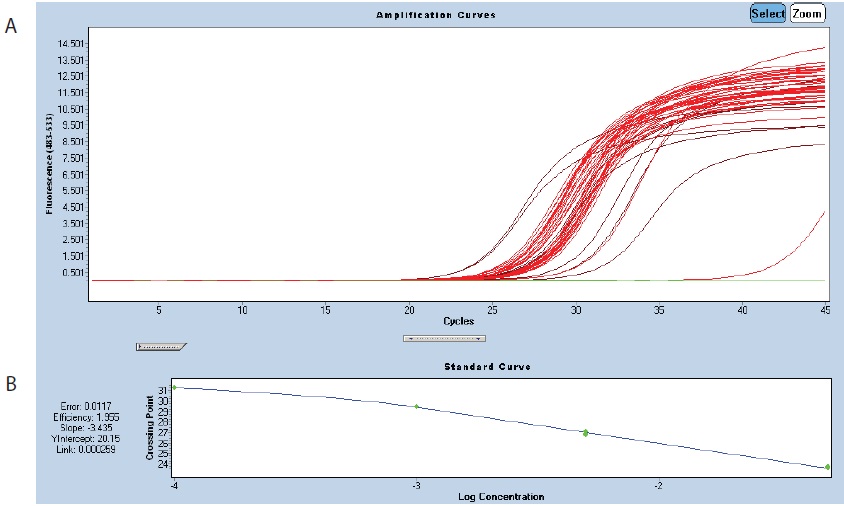
Figure 2. Amplification of rpl2. A. Amplification curves of the standard dilutions of the control sample (brown curves) and 14 other samples (red curves). In green, the negative control. B. Standard curve showing a primer efficiency close to 2. - Quantitative RT-PCR is conducted in 384-well plates (LightCycler 480 Multiwell plate) with a LightCycler 480. A reaction volume of 5 µl is sufficient as mitochondrial transcripts are highly expressed. For the qPCR run, the plates are sealed with the dedicated LightCycler 480 Seal Foil. Keep plates on ice and in the dark until the run.
- Each 5-µl reaction contains 0.5 µl of cDNA dilution, 2.5 µl of LightCycler 480 SYBR I Master mix (comprising DNA polymerase, buffer and DNA double-strand specific SYBR Green I dye) and 2 µl of 2.5 µM primer mix (1.25 µM each). The details of the serial dilutions and reactions mixes are given in Table 3. The plate setup shown in Table 2 allows the analysis of 6 samples (3x 384-well plates for each assay), one sample being used as a control for quantitation (3 biological repeats of wild-type and 3 of mutants for example). Three technical repeats per point (1/200 dilution) are necessary.
- A standard curve is performed for each primer pair, using one of the samples as a control. This cDNA is diluted (1/20, 1/200, 1/1,000, 1/5,000) and the remaining samples at 1/200 only). The primer efficiency is calculated for each pair of primers by the software when doing the standard curve.
- Primer pairs for the mitochondrial transcriptome assay are described in Table 1, as well as the primers for reference genes that will be used for normalisation of the results. It is very difficult to find reference genes whose expression will not vary between samples; it is a matter of trying several, picking the most suitable ones within an experiment and combining them. Alternatively, normalisation can be made to the median of mitochondrial transcripts.
- The primers used for the splicing assay (i.e. for quantifying the splicing of mitochondrial mRNAs) were designed to amplify 100-200 bp regions spanning intron-exon junctions (unspliced forms) or spanning splice junctions (spliced forms) of each gene (Table 1).
- The cDNAs can be tested by a first round of qPCR using one pair of nuclear primers and one pair of mitochondrial primers from the assay (see Table 1). This will allow you to adjust the cDNA concentrations (by comparing the crossing points (Cp) for all samples) before setting up 3 or 6 x 384-well plates (Figure 2).
- Quantitative RT-PCR-time course for pipetting
- Prepare the primer plates
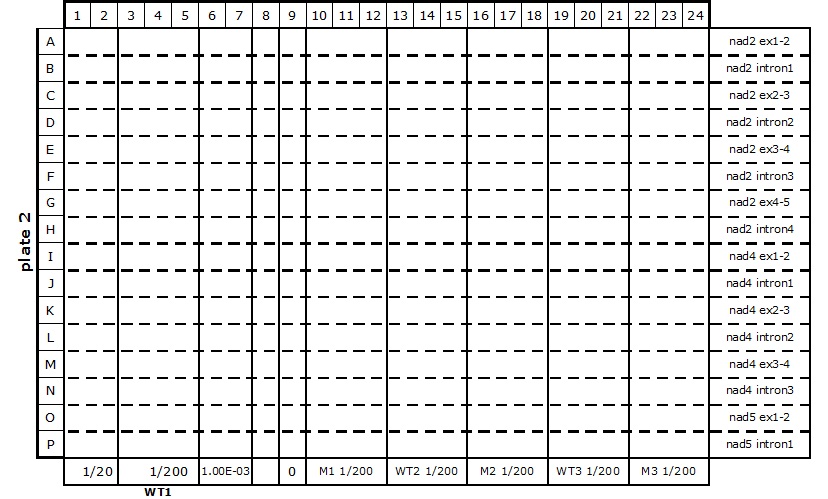
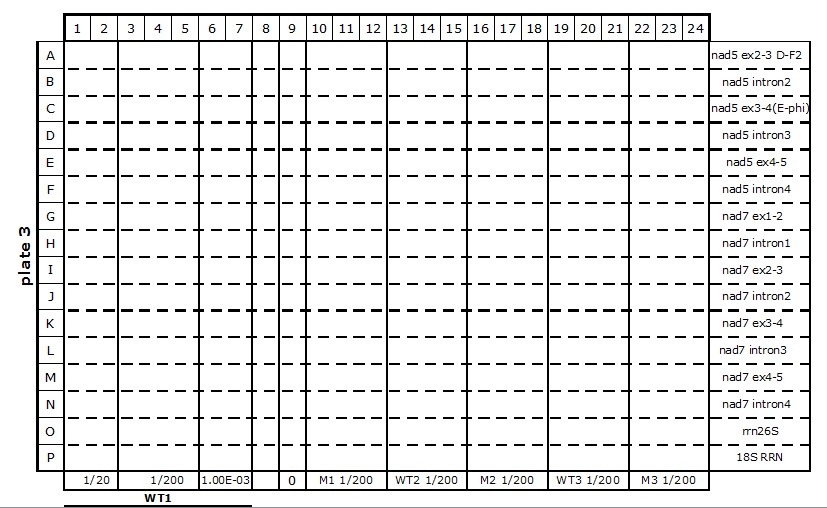
The primers (2 µl at 1.25 µM each) are dispensed into the wells with an automatic dispenser ‘Multipette plus’, one primer pair per horizontal line (see Table 2). All plate set up stages must be done on ice, or in the cold room in case of multiple plate set up, to limit evaporation of the reagents.
This step can be done in advance and the plates (sealed with Adhesive PCR film seals from Thermo Scientific) can be kept at -20 °C.
Table 2. 384-well plate set up for mitochondrial transcriptome and splicing assay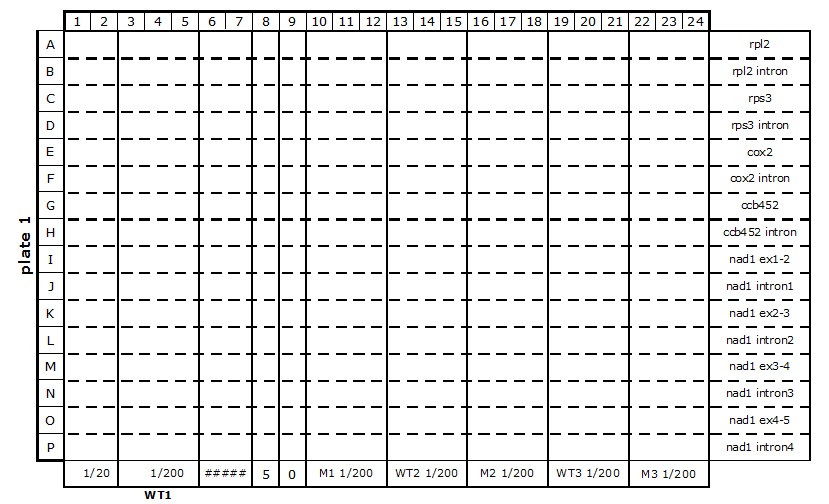
- The reaction mixes are prepared according to Table 3 and kept on ice.
Prepare serial cDNA dilutions and add the exact volume of SYBR Master mix for each dilution. - Add 3 µl of this mix (SYBR Master mix + cDNA) to each well already containing 2 µl of primer mixes at 1.25 µM each.
- Seal and briefly spin the plates, keep them on ice and in the dark.
Table 3. Serial cDNA dilutions and qPCR mixesVolume 52 µl 76 µl 52 µl 26 µl 26 µl 76 µl Dilution 1/20 1/200 1/1,000 1/5,000 0 Mutant/200 Water 57.8 µl 78.8 µl 45.8 µl 20.8 µl 26 75.6 µl cDNA 3.04 µl 0.38 µl 1/20 8.75 µl 1/200 11.44 µl 1/1,000 5.2 µl 1/20 1/200 1/1,000 1/5,000 0 Mutant/200 Master mix 260 µl 380 µl 260 µl 130 µl 130 µl 380 µl cDNA 52 µl 76 µl 52 µl 26 µl 26 µl 76 µl - Perform qPCR run as soon as possible. The thermal cycling program is: 95 °C for 10 min, followed by 45 cycles of 95 °C for 10 sec, 60 °C for 10 sec and 72 °C for 20 sec.
- The plate set-up should be entered in the LC480 software before starting the run, but this can be done later if necessary. Under ‘absolute quantitation’, fill in the ’Subset editor’ with the transcript names and positions in the plate and finally the ‘Sample editor’.
- Prepare the primer plates
Data analysis
The data are analysed using the inbuilt LightCycler 480 software version 1.5. The Second Derivative Maximum Method, which determines the Cp value at the beginning of the log-linear phase of the real-time fluorescence (Luu-The et al., 2005) is fast and easy, but the fit point method can be used too. All transcripts are quantified relative to an internal control sample. For each primer pair, a standard curve is done with the internal control sample. The efficiency of the PCR reaction is checked (optimal efficiency is 2, but is acceptable between 1.6 and 2.4). For the error to be minimal, standard errors should be less than 10 %, but for some primer pairs it is difficult to achieve.
The figures are then copied into a spreadsheet and calculations are done as follows:
- A normalisation step is necessary: For this, use either the geometric mean of 2 to 3 suitable reference genes or normalise to the median of all mitochondrial transcripts. It is always best to try several ways of doing it.
- Normalise each sample to its control, i.e., calculate ratio of expression of mutant M1 to WT1, M2/WT2, M3/WT3 or whatever comparison you need to do.
- Draw a first graph of transcript accumulation in mutant/WT. Showing the results with a log2 scale can be easier in some cases (see Figure 3).
- Calculate an average and a standard deviation (SD) between biological repeats. If the interval between the average log2 ratio + SD and average log2 ratio-SD does not contain zero, then the change is considered significant.
- For the splicing assay, the results are best presented as the log2 ratio of (spliced mRNA in the mutant to spliced mRNA in the WT) to (unspliced mRNA in the mutant to the unspliced mRNA in the WT) (Figure 3A). Nevertheless, to be sure what you see is the effect of a true splicing defect, you must check that the accumulation of spliced mRNA is down in the mutant and that of the unspliced mRNA is up in the mutant.
- Results can be confirmed by northern blots if necessary.
Representative data
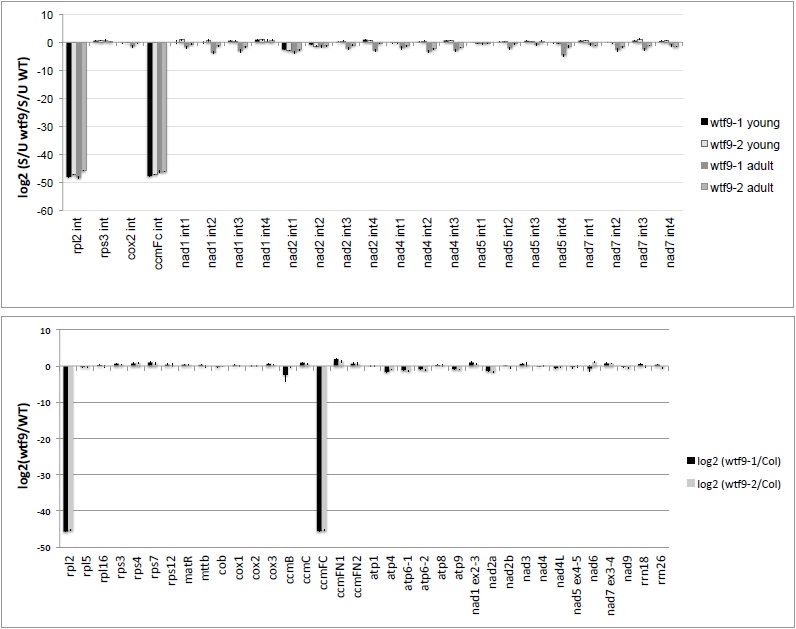
Figure 3. Representative data. A. Panel A presents the qRT-PCR mitochondrial splicing assay for the mutant wtf9 (Francs-Small et al., 2012), which has a defect in rpl2 and ccmFc transcript splicing. The defects are very obvious when the data is represented as a log2 value of the relative quantities of spliced to unspliced forms of each transcript. B. Panel B shows the transcriptome data, where mRNA levels are expressed as a ratio of transcript levels in mutants compared with levels in WT Col-0 plants.
Notes
These assays are generally very reproducible and reliable due to the abundance of mitochondrial transcripts in plant tissues. Of course, in some mutants, some transcripts are expressed at significantly different levels compared to WT but this is what the essay is designed to detect.
Acknowledgments
This protocol was originally developed by Etiennne Delannoy and Andéol Falcon de Longevialle for work supported by the Australian Research Council Centre of Excellence grant CE0561495 (de Longevialle et al., 2007; Koprivova et al., 2010; Kühn et al., 2009). Some primer pairs have been modified or added by C. Colas des Francs-Small, in particular for the study of nad5 splicing (Colas des Francs-Small et al., 2014).
References
- Colas des Francs-Small, C., Falcon de Longevialle, A., Li, Y., Lowe, E., Tanz, S. K., Smith, C., Bevan, M. W. and Small, I. (2014). The pentatricopeptide repeat proteins TANG2 and ORGANELLE TRANSCRIPT PROCESSING439 are involved in the splicing of the multipartite nad5 transcript encoding a subunit of mitochondrial complex I. Plant Physiol 165(4): 1409-1416.
- de Longevialle, A. F., Meyer, E. H., Andres, C., Taylor, N. L., Lurin, C., Millar, A. H. and Small, I. D. (2007). The pentatricopeptide repeat gene OTP43 is required for trans-splicing of the mitochondrial nad1 Intron 1 in Arabidopsis thaliana. Plant Cell 19(10): 3256-3265.
- Francs-Small, C. C., Kroeger, T., Zmudjak, M., Ostersetzer-Biran, O., Rahimi, N., Small, I. and Barkan, A. (2012). A PORR domain protein required for rpl2 and ccmF(C) intron splicing and for the biogenesis of c-type cytochromes in Arabidopsis mitochondria. Plant J 69(6): 996-1005.
- Koprivova, A., des Francs-Small, C. C., Calder, G., Mugford, S. T., Tanz, S., Lee, B. R., Zechmann, B., Small, I. and Kopriva, S. (2010). Identification of a pentatricopeptide repeat protein implicated in splicing of intron 1 of mitochondrial nad7 transcripts. J Biol Chem 285(42): 32192-32199.
- Kühn, K., Richter, U., Meyer, E. H., Delannoy, E., Falcon de Longevialle, A. , O'Toole, N., Börner, T., Millar, A. H., Small, I. D. and Whelan, J. (2009). Phage-type RNA polymerase RPOTmp performs gene-specific transcription in mitochondria of Arabidopsis thaliana. Plant Cell 21(9): 2762-2779.
- Luu-The, V., Paquet, N., Calvo, E. and Cumps, J. (2005). Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. Biotechniques 38(2): 287-293.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Delannoy, E., Falcon de Longevialle, A. and Francs-Small, C. C. D. (2015). Mitochondrial RNA Transcript Analysis Assay of Arabidopsis Leaf Tissues. Bio-protocol 5(20): e1620. DOI: 10.21769/BioProtoc.1620.
- Colas des Francs-Small, C., Falcon de Longevialle, A., Li, Y., Lowe, E., Tanz, S. K., Smith, C., Bevan, M. W. and Small, I. (2014). The pentatricopeptide repeat proteins TANG2 and ORGANELLE TRANSCRIPT PROCESSING439 are involved in the splicing of the multipartite nad5 transcript encoding a subunit of mitochondrial complex I. Plant Physiol 165(4): 1409-1416.
Category
Plant Science > Plant molecular biology > RNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.