- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Human, Bacterial and Fungal Amplicon Collection and Processing for Sequencing
Published: Vol 5, Iss 10, May 20, 2015 DOI: 10.21769/BioProtoc.1477 Views: 13708
Reviewed by: Aksiniya AsenovaEmily CopeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Sequencing taxonomic marker genes is a powerful tool to interrogate the composition of microbial communities. For example, bacterial and fungal community composition can be evaluated in parallel using the 16S ribosomal RNA gene for bacteria or the internal transcribed spacer region in fungi. These are conserved regions that are universal to a taxonomic clade, yet have undergone some degree of evolution such that different lineages can be differentiated. Conserved regions are used for design of universal priming sites that allow amplification of the marker gene out of a mixed microbial community. Here, we describe our standard operating procedure to collect and sequence 16S rRNA and ITS1 amplicons from human skin. We use the 16S rRNA V1-V3 region for skin samples, as it has greater power for classifying common staphylococci in the skin. This protocol is adapted for 454 pyrosequencing of amplicons.
Keywords: MicrobiomeMaterials and Reagents
- Sample collection and DNA extraction
- Catch-All sample collection swabs (Epicentre, catalog number: QEC89100 )
- MasterPure yeast DNA purification kit (Epicentre, catalog number: MPY80200 )
- Yeast cell lysis buffer (Epicentre, catalog number: MPY80200)
- ReadyLyse (Epicentre, catalog number: R1810M )
- Promega DNA IQ Spin baskets (Promega Corporation, catalog number: U1221 )
- Stainless steel beads, 5mm (QIAGEN, catalog number: 69989 )
- MoBio PCR water (MoBio, catalog number: 17000-10 )
- 100% (200 proof) ethanol (Warner-Graham Company, catalog number: 64-17-5 )
- 70% ethanol made from 350 ml of 100% ethyl alcohol and 150 ml of MoBio water
- PureLink Genomic DNA Mini Kit (Life Technologies, InvitrogenTM, catalog number: K182002 )
- Catch-All sample collection swabs (Epicentre, catalog number: QEC89100 )
- PCR amplification
- Accuprime Taq polymerase HiFi (Life Technologies, InvitrogenTM, catalog number: 12346-086 )
- Forward and reverse primers with designated barcodes (IDT custom order)
- MinElute PCR purification kit (QIAGEN, catalog number: 28006 )
- QuantIT ds DNA assay, high sensitivity (Life Technologies, InvitrogenTM, catalog number: P7589 )
- Ampure (SPRI) beads, 60 ml kit (Agencourt, catalog number: A63881 )
- TE (pH 8.0) (Life Technologies, catalog number: AM9849 )
- Accuprime Taq polymerase HiFi (Life Technologies, InvitrogenTM, catalog number: 12346-086 )
Equipment
- 2.0 ml Safe-Lock Biopur individually sealed tubes (Eppendorf, catalog number: 0030 121.597 )
- Sterile scissors (VWR, catalog number: 82027-594 )
- Sterile tweezers (VWR, catalog number: 231-SA-SE )
- Safe-Lock PCR Biopur tube (Eppendorf, catalog number: 0030 123.344 )
- 15 and 50 ml conical tubes (Falcon, catalog numbers: 14-959-49D and 14-432-22 )
- 96-well thermocycler plates (USA Scientific, catalog number: 1402-9596 )
- Clear adhesive plate seals (USA Scientific, catalog number: 2978-2100 )
- Foil plate seals (USA Scientific, catalog number: 2923-0110 )
- Reagent reservoirs (USA Scientific, catalog number: 2320-2620 )
- Heated shaking block that holds Eppendorf tubes (Eppendorf Thermomixer C)
- Bead beater (Qiagen TissueLyser II, catalog number: 85300 )
- Bead beater adapters (Qiagen TissueLyser Adapter Sets, catalog number: 69982 )
- UV crosslinker (UVP, catalog number: CL-1000 )
- Microcentrifuge (Eppendorf, Centrifuge 5415D )
- Thermocycler (Applied Biosystems Veriti)
- Plate centrifuge (Beckman Coulter, Allegra 6KR)
- Fluorometer plate reader (Thermo Scientific, catalog number: 5210450 )
- Multi-channel pipettes (pre- and post-PCR designated)
- 96- well microplates for fluorescence-based assays (Life Technologies, catalog number: M33089 )
- Qubit 2.0 Fluorometer (Life Technologies, InvitrogenTM, catalog number: Q32866 )
- Qubit 2.0 Quantitation Starter Kit (Life Technologies, InvitrogenTM, catalog number: Q32871 )
Procedure
- Sample collection
- Add 100 μl of yeast cell lysis buffer to a 2 ml safe-lock tube.
- Open the swab pack, pre-moisten swab in lysis buffer, and then swab the selected skin site with the swab for 10 sec. For high-resolution skin sampling, a 4 cm2 skin area is swabbed. Then return the swab to lysis buffer and with sterile scissors, cut off the swab so that the top is even with the swab stem so that it will fit in the tube. If the swab is cut too short, recovery during the DNA extraction step is more difficult. Collect a negative control by waving a pre-moistened swab in the air, then placing into the lysis buffer.
- Samples in lysis buffer can be stored at -80 °C prior to processing. While it is unknown whether samples are stable indefinitely, good results have been obtained from samples frozen for several years.
- Add 100 μl of yeast cell lysis buffer to a 2 ml safe-lock tube.
- DNA extraction
- Clean bench and centrifuge with 70% ethanol and 10% bleach. UV-treat pipettes and tube racks for 30 min prior to use.
- Thaw tubes for 10 min at 37 °C on heated shaking block.
- Short spin tubes, then open and add 1 μl of ReadyLyse and 250 μl of Yeast Cell Lysis Solution.
- Incubate tubes for 60 min at 37 °C on heated shaking block.
- During this time, UV-treat for 30 min an appropriate number of Promega IQ Spin baskets and stainless steel beads, for example by placing them in a sterile, covered petri dish. Also UV-treat a pair of tweezers.
- After incubation, short spin tubes.
- Pick up a Promega basket with tweezers one hand, open a tube with the other hand. Take the basket with the other hand and then use the tweezers to pick up the tip of the swab. While holding the swab, place the basket into the tube, then replace the swab into the tube+basket. Between samples, wipe the tweezers well with a paper towel soaked with 70% ethanol.
- Place the open tube + basket + swab into a centrifuge and short spin to collect excess buffer. Remove basket + swab and discard.
- With tweezers wiped down with 70% ethanol, open the tube and add two 5-mm stainless steel beads to each sample taking care not to touch the tweezers to the tube. Close tubes and load tubes into the QIAGEN bead beater adaptor and balance. Beat for 2 min at 30 Hz.
- Removed tubes and incubate for 30 min at 65 °C on heated shaking block.
- Place on ice for 5 min. Short spin tubes.
- Add 150 μl Epicentre MPC Reagent. Vortex tube 10 sec, then spin 10 min. In the meanwhile, label a fresh set of 2 ml Eppendorf tubes.
- Transfer the supernatant, being careful not to disturb the pellet at the bottom of the tube, to the fresh tube and add an equal volume of 100% EtOH. Mix by inverting several times.
- Set up PureLink Genomic DNA Mini Kit genomic DNA extraction columns with their collection tubes according to manufacturer’s instructions. Apply sample to the extraction column. Spin at maximum speed for 1 min. Remove flow-through and repeat if sample remains. Place column into a fresh collection tube.
- Wash column with 500 μl Buffer 1, spin at maximum speed for 1 min, discard flow-through, and place column into a fresh collection tube.
- Wash column with 500 μl Buffer 2. Spin at maximum speed for 1 min, discard flow-through, and then spin at maximum speed for 3 min to dry the column.
- Discard flow-through and place column in clean safe-lock PCR tube. Add 25 μl ultrapure PCR water and let stand for 3 min. Spin at maximum speed for 30 sec to elute. Discard column and store eluate at -20 °C.
- Resultant genomic DNA is typically quantified according to manufacturer’s instructions with Qubit fluorometric quantitation. Regardless of concentration, 2 μl per sample is used for subsequent amplification.
- Clean bench and centrifuge with 70% ethanol and 10% bleach. UV-treat pipettes and tube racks for 30 min prior to use.
- 16S amplification of V1-V3 region for 454 sequencing
- Here, 2 μl of eluate per reaction is used, and reactions are performed in duplicate and combined prior to purification. The concentration of the eluate is highly variable depending on sample origin. For example, low abundance skin sites can yield under 1 ng/μl, while high abundance skin sites can yield greater than 50 ng/μl.
Primer V1_27F: 5’-AGAG TTTGATCCTGGCTCAG-3’
Primer V3_534R w/ barcodes: 5’-ATTACCGCGGCTGCTGG-3’
Primers to other regions, and sample barcodes, are available online at http://www.hmpdacc.org.
- To create the premix for two 96-well plates, combine buffer, Taq, forward primer, and water into a 15 ml conical tube and vortex to mix. Keep on ice. All amounts are in μl.
1 reaction
110x reactions
10x Accuprime buffer II
2
220
Accuprime taq
0.15
16.5
100 μM Primer V1_27F
0.05
2.2
2 μM Primer V3_534R with barcode
2
-
DNA eluate
2
-
Water (for PCR)
13.8
1519.1
- Aliquot 23 μl of premix into each well of two 96-well plates.
- Add the barcoded reverse primer to each well.
- Add DNA to each well.
- Seal plates with thermofoil, and spin down.
- Run the following PCR program:
95 °C, 2 min
30 cycles of:
95 °C, 20 sec
56 °C, 30 sec
72 °C, 5 min
Then,
4 °C, forever
- Here, 2 μl of eluate per reaction is used, and reactions are performed in duplicate and combined prior to purification. The concentration of the eluate is highly variable depending on sample origin. For example, low abundance skin sites can yield under 1 ng/μl, while high abundance skin sites can yield greater than 50 ng/μl.
- ITS1 amplification for 454 sequencing
- Here, 4 μl of eluate per reaction is used, and reactions are performed in duplicate and combined prior to purification.
Primer ITS1F: 5’-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGGTAAAAG TCGTAACAAGGTTTC-3’
Primer ITS1R with barcodes: 5’- GTTCAAAGAYTCGATGATTCAC-3’
- To create the premix for two 96-well plates, combine buffer, Taq, forward primer, and water into a 15 ml concial tube and vortex to mix. Keep on ice. All amounts are in μl.
1 reaction
110x reactions
10x Accuprime buffer II
2.5
275
Accuprime taq
0.2
22
100 μM Primer ITSF
0.1
11
Water (for PCR)
16.2
2002
2 μM Primer ITS1R with barcode
2
-
DNA eluate
4
-
- Aliquot 19 μl of premix into each well of two 96-well plates.
- Add the barcoded reverse primer to each well.
- Add DNA to each well.
- Seal plates with foil plate seals, and spin down.
- Run the following PCR program:
95 °C, 2 min
32 cycles of:
95 °C, 30 sec
50 °C, 30 sec
72 °C, 2 min
Then,
72 °C, 5 min
4 °C, forever
- Here, 4 μl of eluate per reaction is used, and reactions are performed in duplicate and combined prior to purification.
- PCR Clean-up with Agencourt SPRI kit
- Spin down plates, then combine V1-V3 reactions into one plate, and ITS reactions into one plate.
- Add 72 μl Agencourt Beads to each well, pipetting up and down 10x.
- Incubate at room temperature for 5 min to bind DNA to beads.
- Place plate on magnet for 2 min to separate beads from solution.
- Keeping the plate on the magnet, aspirate cleared solution from reaction plate (take care to avoid aspirating beads on the bottom of the wells), and discard.
- Keeping the plate on the magnet, add 200 μl 70% ethanol to each well. Aspirate. Repeat.
- Keep the plate open for 5 min to dry beads.
- Remove the plate off the magnet and add 25 μl TE, pH 8.0 to the plate. Allow to elute for 1 min.
- Place plate back on magnet and allow beads to separate for 1 min.
- Transfer eluate to a new plate.
- Spin down plates, then combine V1-V3 reactions into one plate, and ITS reactions into one plate.
- PCR product quantification using QuantIT dsDNA high-sensitivity assay kit
- Bring kit components to room temperature.
- For two plates, mix 40 ml of QuantIT buffer with 200 μl of QuantIT reagent in a 50 ml conical tube.
- Aliquot 200 μl of buffer-reagent mix to each well of two 96 well microplates for fluorescence-based assays.
- Add 10 μl of each standard from the kit (8 total standards at 0, 0.5, 1, 2, 4, 6, 8, and 10 ng/μl) to first two columns on each plate. Standards are read in duplicate and samples a single time.
- Add 2 μl of PCR products to wells, leaving a column of blanks between the standards and samples.
- Including a 10 sec shake step, measure fluorescence on Thermo plate reader with filter pair 485/538.
- Generate standard curve from standards, subtracting background.
- Calculate PCR product concentration from standard curve and equation.
- Bring kit components to room temperature.
- PCR product pooling and purification
- Pool approximately equal amounts of each PCR product into a single Eppendorf tube. The target ng per sample is determined by the lowest concentration sample (excluding controls). For the lowest concentration sample(s), add the entire PCR product, and for those with higher amounts, add an approximately equivalent amount.
- Purify pool with QIAGEN minElute column according to manufacturer’s instructions. Briefly, add 5 volumes of buffer PB to 1 volume of the combined PCR pool.
- Apply the sample to the MinElute column that has been placed in a 2 ml collection tube.
- To bind DNA, centrifuge 30 sec at maximum speed. Discard flowthrough and place the column back in the same tube.
- Wash with 750 μl buffer PE, discard flowthrough, then centrifuge for an additional 1 min.
- Place the column into a fresh tube and elute in 30 μl TE (pH 8.0). This final product represents an approximately equivalent concentration of each of the 96 input samples. To ensure quality, one may then quantitate the pooled library according to manufacturer’s instructions with Qubit fluorometric quantitation, or by running 2 μl on an electrophoretic agarose gel or equivalent. For the V1-V3 amplicon, the product size should fall between 450-550 bp.
- Single, pooled amplicon libraries in an Eppendorf tubes can then be sent to a sequencing facility for sequencing on a 454 GS FLX (Roche, http://454.com/products/gs-flx-system/index.asp) instrument using titanium chemistry.
- Pool approximately equal amounts of each PCR product into a single Eppendorf tube. The target ng per sample is determined by the lowest concentration sample (excluding controls). For the lowest concentration sample(s), add the entire PCR product, and for those with higher amounts, add an approximately equivalent amount.
Representative data
- For sequence processing, we recommend using the mothur software (http://www.mothur.org) together with the 454 standard operating protocol written by Pat Schloss (http://www.mothur.org/wiki/454_SOP):
- In summary, the first purpose of sequence processing is deconvolutes the raw 454 data into individual samples and then to remove poor quality sequence arising from technical artifacts produced by PCR or 454 sequencing error. The second step of sequence processing is then to make sense of the amplicon sequences. For studies based on 16S rRNA amplicon sequencing, sequences are processed into operational taxonomic units (OTUs) that represent a bacterial or archaeal ‘species’. For fungal studies based on the ITS, we prefer to classify sequences into ‘phylotypes’, as sequence distance that is the basis of the formation of OTUs is very variable for the ITS, which is under very different selective pressure than the 16S rRNA, and depends on the fungal taxon. For species level classification of genera of interest, we created custom taxonomic references using the pplacer algorithm, written by Eric Matsen (http://matsen.fhcrc.org/pplacer). For example, to speciate staphylococcal sequences, 16S full-length rRNA sequences belonging to different staphylococcal species can be downloaded from Genbank or RDP (http://rdp.cme.msu.edu) for construction of the phylogenetic tree. It is important to use high quality (e.g., type strains, fully sequenced genomes) to construct the phylogenetic model.
- We then use the R software to post-process files generated by mothur. For example, to generate plots of the relative abundances between species, we use the *.tax.summary file generated by the classify.seqs() or the classify.otu() command in mothur (Figure 1). A sample R script with sample data are provided at https://github.com/julia0h/amplicon_bioprotocol.git in the plottingTaxonomy/ folder.
- For examining alpha diversity in the sample, we typically use the Shannon diversity index, Inverse Simpson, or species observed from the summary.single() command in mothur (Figure 2). Sample scripts are provided in plottingDiversity/.
- Finally, for comparing samples with a distance metric like the Yue-Clayton theta index to generate principal coordinate analysis plots, we use the output of the dist.shared() and pcoa() commands (Figure 3). Sample scripts are provided in plottingPCOA/.
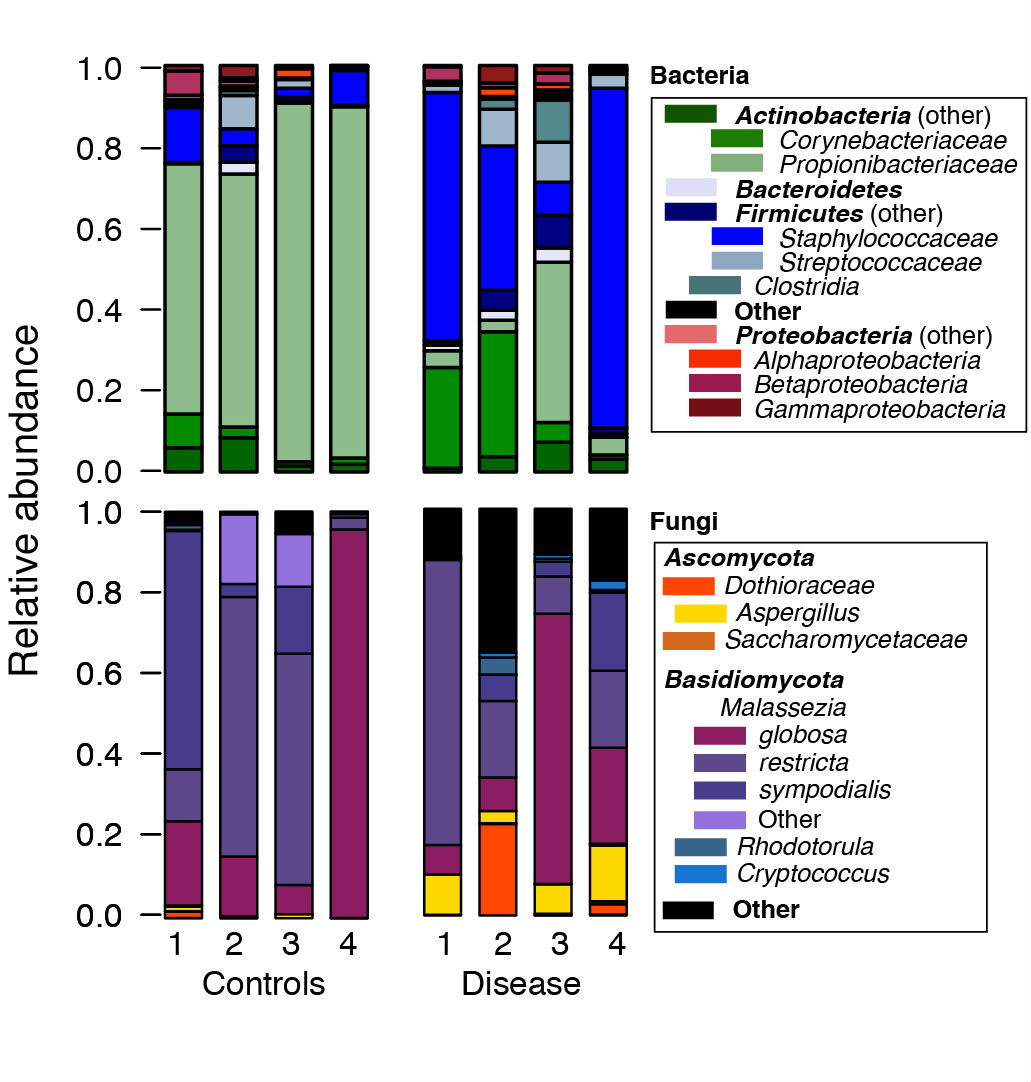
Figure 1. Example relative abundance plots of a control vs disease group
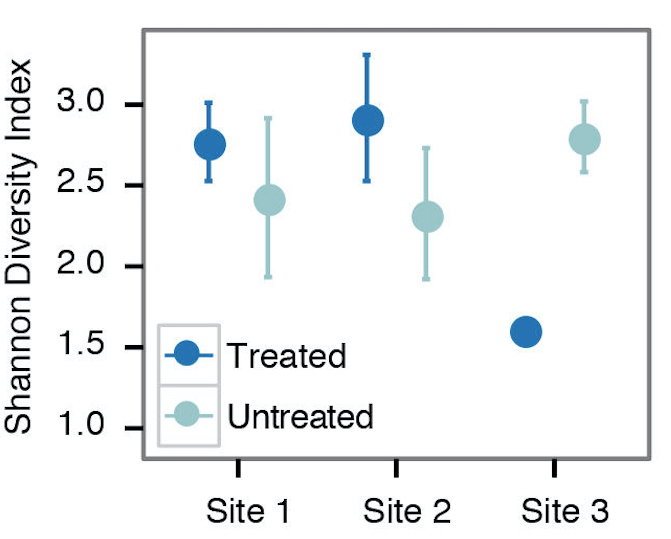
Figure 2. Example alpha diversity analysis looking at the differences in Shannon diversity at 3 different sites that are/are not treated
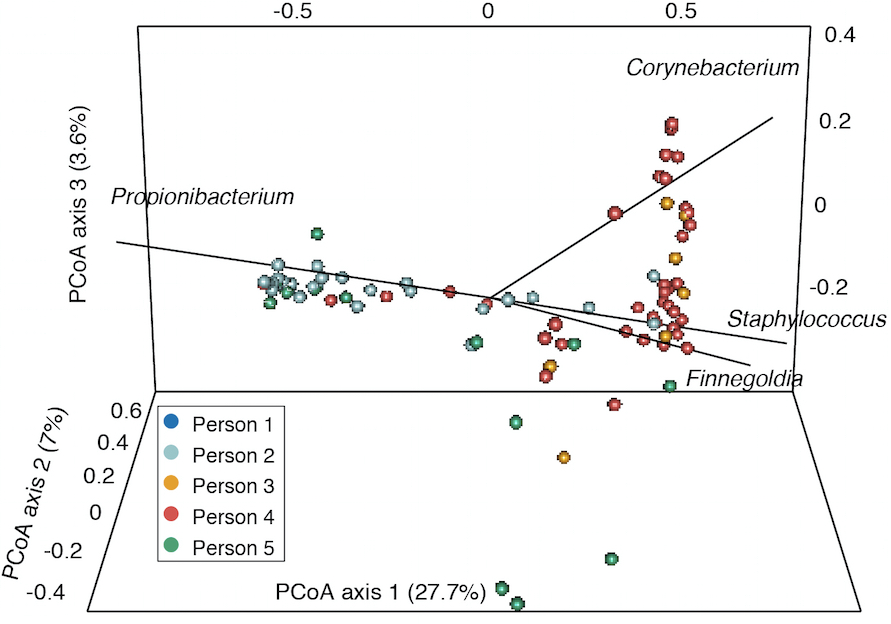
Figure 3. Example principle coordinates analysis based on Yue-Clayton theta values that show the similarity between samples from different individuals. Taxa contributing significantly to variation between samples are shown with lines.
Acknowledgments
This protocol is based on the data generated and analyses performed in Oh et al. (2013). Skin sampling procedures were designed by Dr. Heidi Kong, and DNA extraction and amplification procedures by Clayton Deming, Cynthia Ng, and Elizabeth Grice at the National Institutes of Health.
References
- Oh, J., Freeman, A. F., Program, N. C. S., Park, M., Sokolic, R., Candotti, F., Holland, S. M., Segre, J. A. and Kong, H. H. (2013). The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res 23(12): 2103-2114.
- Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. J. and Weber, C. F. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23): 7537-7541.
- Schloss, P. D., Gevers, D. and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6(12): e27310.
- Matsen, F. A., Kodner, R. B. and Armbrust, E. V. (2010). pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11: 538.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Oh, J. (2015). Human, Bacterial and Fungal Amplicon Collection and Processing for Sequencing. Bio-protocol 5(10): e1477. DOI: 10.21769/BioProtoc.1477.
Category
Microbiology > Microbial genetics > DNA > DNA sequencing
Microbiology > Microbe-host interactions > In vivo model > Mammal
Systems Biology > Genomics > Sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.