- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Chemiluminescence Based Receptor-ligand Binding Assay Using Peptide Ligands with an Acridinium Ester Label
Published: Vol 5, Iss 6, Mar 20, 2015 DOI: 10.21769/BioProtoc.1422 Views: 11833
Reviewed by: Tie LiuMasahiro Morita
Original research article
The authors used this protocol in:

Jun 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Studying the biochemical interaction of ligands with their corresponding receptors requires highly sensitive detection and monitoring of the bound ligand. Classically, radioactively labelled ligands have been widely used as highly sensitive tools for such binding measurements. Disadvantages of radiolabelling include instability of products, high costs and risks of working with radioactivity. Thus, assays using chemiluminescent probes offer convenient, highly sensitive alternatives. Here we suggest acridinium esters as suitable conjugates to label ligands of interest. Chemical oxidation of acridinium esters triggers chemiluminescence, allowing quantitation of this compound down to amol concentrations in standard luminometers. The first report about acridinium esters in immunoassays date back to 1983 (Weeks et al., 1983) and demonstrated the ability to conjugate acridinium to peptides, followed by using such peptides to measure receptor – peptide ligand interactions (Joss and Towbin, 1994).
Recently, this binding assay was adapted for studying derivatives of the plant peptide IDA (INFLORESCENCE DEFICIENT IN ABSCISSION) and their interaction with the corresponding receptor HSL2 (HAESA-LIKE 2) was reported (Butenko et al., 2014). Here we describe how this sensitive, nonradioactive binding approach can be used to reveal receptor-ligand binding in plant material.
Materials and Reagents
- Plant material harboring the receptor of interest in an immobilized form
Suggested plant materials could be: Intact cells from suspension cultures, ground plant tissue or isolated receptors immobilized on immunoprecipitation beads.
Notes:
- The receptor must be immobilized (on cells, cell debris, IP-beads) in order to allow efficient washing to remove unbound ligand without washing away the receptor molecules.
- Levels of some receptors are exceedingly low. Therefore, overexpression, e.g. by transient expression in Nicotiana benthamiana (N. benthamiana) leaves (Li, 2011), may be required to obtain sufficient binding sites.
- Use a negative control, same material lacking a functional receptor, whenever possible.
- The receptor must be immobilized (on cells, cell debris, IP-beads) in order to allow efficient washing to remove unbound ligand without washing away the receptor molecules.
- MES (2-ethanesulfonic acid)
- NaCl
- DTT
- 33 µl proteinase Inhibitor (Sigma-Aldrich, catalog number: P9599 )
- 5 mM citric acid
- 0.03 % H2O2 in 100 mM NaOH (prepare fresh from a 30% H2O2 stock)
- Appropriate binding buffer (see Recipes)
Equipment
- Mortar and pestle
- Table top centrifuge
- Luminometer (e.g. single tube machines like FB 12, Berthold technologies, that allows injection)
- Acridinium-labeled peptide
Important: The modified peptide should be tested for biological activity in your usually used bio-assay of choice and should not deviate much from the activity of the unmodified peptide.
- Unlabeled peptide (synthetic unlabeled peptides can be ordered by e.g. Biomatik)
- Peptides and conjugation with acridinium
Note: Peptides can be ordered (on resin) from a company of choice or synthesized using solid-phase technology with Fmoc-protected amino acids. Acridinium esters can be conjugated to the N-terminal amino groups of the peptides on resin by coupling with N-hydroxysuccinimide activated acridinium esters (Cayman) before deprotection and purification of the peptides via HPLC.
Procedure
Note: Here, the procedure is described for using frozen, ground leaf tissue but the method should be easily adaptable for other materials.
- Shock-freeze leaf tissue (age does not matter, however, no senescence symptoms should be visible) or any material of choice (cell suspension culturue, e.g., N. benthamiana leaves in liquid nitrogen.
Note: If using transient expression by agroinfiltration, or other transient expression methods, the time until your protein is expressed at high levels might vary, depending on the protein, promoter etc. Make sure to use a tag in order for expression level to be detected.
- Grind tissue in liquid N2 to a fine powder with mortar and pestle.
- Using a pre-cooled spatula, place the right amount of tissue in a pre-cooled 1.5 ml reaction tube. We use 500 mg tissue (frozen) for 10-12 individual samples/measurements (~ 40-50 mg per sample).
- Suspend in binding buffer (700-1,000 µl).
- Centrifuge suspension to create a pellet (13,000 x g, 1 min, 4 °C).
- Resuspend the pellet in binding buffer (500 mg tissue/ml binding buffer).
- Aliquot in 1.5 ml reaction tubes, 80 μl in each tube (n ≥ 3 per treatment). Cut off the tip of the pipette helps to avoid clogging of tissue at the tip and to get an even distribution of tissue in the samples.
- Add 20 μl of a solution containing the desired combination of acridinium labeled peptide and unlabeled peptide (peptides should be solved/diluted in binding buffer) to reach 100 μl.
Note: Use a final concentration of 0.1 to 10 nM acridinium labeled peptide in your 100 µl sample. Run replicates (n ≥ 3) and samples containing an excess (>1,000-fold) of unlabeled peptide to determine non-specific binding (e.g. 1 nM Acridinium-peptide plus 1 µM unlabeled peptide as competitor).
- 20 min incubation (4 °C).
Note: Reaching binding equilibrium/saturation may differ and experiments to optimize may be necessary. When no specific binding could be detected, parameters such as incubation time or temperature could be increased.
- Sediment the sample by centrifugation (13,000 x g, 1 min, 4 °C), pipette off and discard the supernatant.
- Wash the pellet 2-3 times by resuspension in 1 ml binding buffer and centrifugation (13,000 x g, 1 min, 4 °C).
Note: Keep the total time required for these washing steps at a minimum! Depending on their dissociation rates, prolonged washing may lead to considerable loss of ligand binding.
- Resuspend the final pellet in 100 μl 5 mM citric acid.
- Place tube in the luminometer.
- Start the software and the program so it is ready to start collecting light immediately after induction with the NaOH/H2O2 solution.
Note: Depending on your luminometer/software, you should monitor emitted light in a continuous mode or, if not possible in shortest possible intervals (e.g. each 0.2 sec or 0.5 sec) over time.
- Inject 100 μl of 100 mM NaOH containing 0.03% H2O2 (induction of luminescence) into the tube/cuvette containing your sample.
- Collect data for 10-30 sec.
Note: The oxidation and light emission proceeds rapidly and should lead to a short flash of light (Joss and Towbin, 1994; Butenko et al., 2014). However, assay conditions and plant material may slow down the reaction. Nevertheless, integration time of 10-30 sec should be sufficient to collect > 90 % of the light emitted (Figure 1).
Representative data
Integrals of light emission can used to quantify the amount of ligand bound by comparison to an acridinium-ester standard curve.
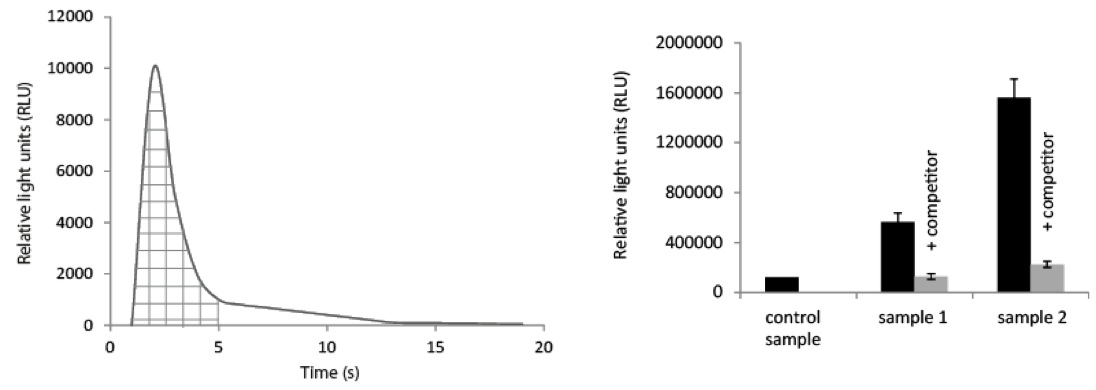
Figure 1. Data set handling. Left panel: The collected data will give you a diagram showing the light (as RLU) collected over the time course of the experiments (left panel). By collecting the data points during the flash peak (shaded area in the figure) and integrating them, the data can be transferred to a diagram for comparison of the samples (to the right). Right panel: Obtained values (as described above) derived from different samples; control sample = plant material without receptor (e.g. from mutant plant, or untransformed plants) incubated with the same amount of Acridinium-labeled peptide and washed the same way as sample 1 and 2, respectively. Sample 1 and 2: grinded plant material with receptor was incubated with Acridinium-labeled peptide alone (black column) or with unlabeled peptide (1,000x excess) as competitor (grey column; + competitor). While the grey columns show the background of unspecific binding and the black column shows the total binding of the Acridinium-peptide the difference between the black and grey columns indicate the specific binding of Acridinium labeled peptide to the corresponding receptor within the respective sample(s).
Notes
- Additional notes and troubleshooting
- Standard curve with the particular acridinium-ligand and the photometer in use.
- Controls to test for light leakage and photoemission by plastic tubes etc should be performed.
- Kinetics of light emission should show a flash (~1 sec) and subsequent exponential decrease of light production.
- Integrals of emitted light (e.g. over 30 sec) should show (close to) linear relationship over several logs.
→ A sensitive photometer allows detection of a functional acridinium label down to the low fmol or amol range.
- Controls to test for light leakage and photoemission by plastic tubes etc should be performed.
- Standard curves/values in the presence of the sample material and buffer conditions to be used in the actual binding assays.
- Controls to test for potential photoemission by the biological material (cells, tissue debris, conA beads, IP beads, etc.), buffers, detergents etc. in use should be added.
- Compare kinetics and integrals of light emission for standard concentrations to the values obtained in 1) to determine physical and chemical quenching.
→ Take quenching into account for determination of amounts of acridinium-labeled peptide present in the samples.
Moderate quenching is inherent to these assays but strong quenching, e.g. > 80% light absorbed from leaf tissue or a strongly distorted kinetics of light emission require assay parameters to be re-adjusted. Such quenching could be avoided in a procedure that re-extracts the acridinium-label from the material before measuring.
- Controls to test for potential photoemission by the biological material (cells, tissue debris, conA beads, IP beads, etc.), buffers, detergents etc. in use should be added.
- 'Non-specific' binding.
- Determine non-specific binding of the sample material (cells, tissue debris, conA beads, IP beads etc.) under the buffer-, timing-, washing- conditions to be used in the actual binding assays. Use either equivalent sample material containing no binding sites or add a high excess of non-labeled ligand (e.g. at concentrations ≥ 10 µM).
- Use these types of assays also to evaluate the washing steps (e.g. balance of pmoles of acridinium-labeled ligand added to the assays versus pmoles remaining after washing).
- The amount of non-specific binding depends on the amount of acridinium-labeled ligand applied (not necessarily in a linear way).
→ A certain amount of non-specific binding is inherent to all binding assays. Calculating the molar amounts that bind non-specifically indicate the minimal amounts of binding sites that have to be present in the samples in order to detect significant amounts of 'specific' binding.
If necessary, minimize non-specific binding. This could be done by e.g. reducing the amount of acridinium-labeled ligand, changing sample material, ionic-conditions and buffering, or by addition of blocking substances to saturate unspecific binding sites on surfaces.
- Determine non-specific binding of the sample material (cells, tissue debris, conA beads, IP beads etc.) under the buffer-, timing-, washing- conditions to be used in the actual binding assays. Use either equivalent sample material containing no binding sites or add a high excess of non-labeled ligand (e.g. at concentrations ≥ 10 µM).
- 'Specific' binding = 'total' binding - 'non-specific' binding.
Problems with detecting specific binding might be due to
- Amount of 'non-specific' binding sites > 'specific' binding sites.
- Low number of binding sites present (confirm presence of receptor molecules with immunological methods if possible).
- Low affinity of binding sites for the acridinium-labeled ligand could lead to loss of bound ligand during lengthy washing steps (washing time matters for reversible binding).
- Wrong/suboptimal conditions for binding [pH, ionic conditions, exposure of binding sites (e.g. vesicle formation), time, temperature].
- Amount of 'non-specific' binding sites > 'specific' binding sites.
- Standard curve with the particular acridinium-ligand and the photometer in use.
- Final remarks
The control parameters listed above define and limit the binding assays. Optimizing the experimental setup for studying the interaction between a given ligand and its particular receptor site might provide some repeated attempts. However, measuring of acridinium is fast and easy, therefore, once the setup is optimalized, several measurements and assay conditions can be tested per day.
Recipes
- Appropriate binding buffer
Example of binding buffer used in (Butenko et al., 2014)
25 mM MES (2-ethanesulfonic acid, pH 6.0)
150 mM NaCl
1 mM DTT (do not add DTT if receptors or ligands are sensitive to reducing agents)
33 µl Proteinase Inhibitor mix per g plant material (essential)
Note: The binding buffer might be adapted or modified by using different buffers adjusted to different pH (e.g. 25 mM MES pH 5.5 or 25 mM TRIS pH 7.5), different salt concentrations (reduce non-specific ionic interactions, e.g. 10, 50 or 100 mM NaCl) in order to optimize binding while keeping non-specific binding at a minimum.
Acknowledgments
R.A.’s work was supported by Grant 348256/F20 from the Research Council of Norway; and Grant 216856 from the Research Council of Norway and the Deutscher Akademischer Austausch Dienst. M. A. was supported by the Deutsch Forschungsgemeinschaft (AL1426/1-1). The method was recently described and applied in Butenko et al. (2014).
References
- Butenko, M. A., Wildhagen, M., Albert, M., Jehle, A., Kalbacher, H., Aalen, R. B. and Felix, G. (2014). Tools and strategies to match peptide-ligand receptor pairs. Plant Cell 26(5): 1838-1847.
- Joss, U. R. and Towbin, H. (1994). Acridinium ester labelled cytokines: receptor binding studies with human interleukin-1 alpha, interleukin-1 beta and interferon-gamma. J Biolumin Chemilumin 9(1): 21-28.
- Li, X. (2011). Infiltration of Nicotiana benthamiana protocol for transient expression via Agrobacterium. Bio-protocol Bio101: e95.
- Weeks, I., Beheshti, I., McCapra, F., Campbell, A. K. and Woodhead, J. S. (1983). Acridinium esters as high-specific-activity labels in immunoassay. Clin Chem 29(8): 1474-1479.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wildhagen, M., Butenko, M. A., Aalen, R. B., Felix, G. and Albert, M. (2015). A Chemiluminescence Based Receptor-ligand Binding Assay Using Peptide Ligands with an Acridinium Ester Label. Bio-protocol 5(6): e1422. DOI: 10.21769/BioProtoc.1422.
Category
Plant Science > Plant biochemistry > Protein > Interaction
Plant Science > Plant immunity > Perception and signaling
Biochemistry > Protein > Interaction > Protein-ligand interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.