- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification and Crystallization of Chloromuconolactone Dehalogenase ClcF from Rhodococcus opacus 1CP
Published: Vol 4, Iss 8, Apr 20, 2014 DOI: 10.21769/BioProtoc.1107 Views: 11475
Reviewed by: Fanglian He
Original research article
The authors used this protocol in:

Apr 2013

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The protocol describes the generation of variants of chloromuconolactone dehalogenase from Rhodococcus opacus (R. opacus) 1CP. ClcF is a multimeric protein, which catalyses the dechlorination of 5-chloromuconolactone to cis-dienelactone in the 3-chlorocatecholic acid degradation pathway. The protocol describes the workflow for the purification and subsequent crystallization of the enzyme. The used workflow and the described techniques could be easily adapted to any other protein/enzyme intended to be crystallized by the potential user for subsequent structure determination. The protocol does not involve expensive specialized equipment which allows the use in standard laboratories not specially dedicated to macromolecular crystallography.
Keywords: CrystallisationMaterials and Reagents
- Escherichia coli (E. coli) BL21 (DE3)-CP-RIL (Stratagene, catalog number: 230245 )
- E.coli DH5α (Life Technologies, catalog number: 18258012 )
- wtClcF plasmid (not commerically available)
- Forward Primer (not commerically available)
- Reverse Primer (not commerically available)
- dNTP-Mix (20 mM) (Thermo Fisher Scientific, catalog number: AB-0196
- Pfu-Ultra (2.5 U/µl) (Agilent, catalog number: 600385 )
- DpnI (Thermo Fisher Scientific, catalog number: ER1701 )
- LB medium
- Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Carl Roth, catalog number: CN08.1 )
- Tris base (Carl Roth, catalog number: 5429.1 )
- DNase (Sigma-Aldrich, catalog number: DN25-100MG )
- Ethylene glycol
- MgCl2 (Carl Roth, catalog number: 2189.2 )
- PEG 3350 (Sigma-Aldrich, catalog number: 202440-250G )
- Bis-Tris (Carl Roth, catalog number: 9140.1 )
- Liquid nitrogen
- Lysis buffer/Ion exchange (IEX) buffer A/Hydrophobic interaction chromatography (HIC) buffer A (see Recipes)
- HIC High salt buffer (see Recipes)
- Wash buffer (see Recipes)
- Crystallization buffers (see Recipes)
Equipment
- Standard laboratory equipment
- Nanodrop device (Thermo Fisher Scientific)
- CrystalQuickTM 96 well crystallization plate Greiner 609101(Jena Bioscience, catalog number: CPL-118S )
- Linbro crystallization plate (Jena Bioscience, catalog number: CPL-101S )
- 22 mm circular cover slides (siliconized) (Jena Bioscience, catalog number: CSL-106 )
- Bayer silicone grease (Jena Bioscience, catalog number: CGR-101 )
- Äkta purification system (GE Healthcare)
- HiTrap Q sepharose column (1 ml) (GE Healthcare, catalog number: 17-5053-01 )
- HiTrap phenyl-sepharose (1 ml) (GE Healthcare, catalog number: 17-1351-01 )
- Stereo microscope (Leica Microsystems, model: MZ12 )
- Eppendorf tubes
- Pipetting robot (if available)
- 37 °C incubator
- Centrifuge
- Vivaspin concentrator (MWCO 30 kDa) (EMD Millipore)
Procedure
- Mutation and overexpression
Site directed mutagenesis by whole plasmid PCR was essentially carried out as described by Weiner et al. (1994).
- The mutagenesis-PCR was setup as followed:
1.25 µl wtClcF plasmid (6 kbp) (20 ng/µl)
1.25 µl forward Primer (100 ng/µl or 10 µM) with the respective mutation site
1.25 µl reverse Primer (100 ng/µl or 10 µM) with the respective mutation site
5 µl dNTP-Mix (2.5 mM)
5 µl Pfu-Ultra buffer
1 µl Pfu-Ultra 2.5 U/µl
35.25 µl Water
PCR-Program:
Temperature
Time
95 °C
30 sec
95 °C
30 sec
18 cycles
55 °C
1 min
72 °C
1 min/kbp plasmid
- At the end the reaction was supplemented with 1/10 volume of 10x DpnI buffer and 10 U DpnI were added. The reaction was incubated at 37 °C for 1 h.
- Afterwards 1.25 µl of this mixture was transformed in ultracompetent E. coli DH5α.
- Sequence integrity has to be proved by sequencing.
- For expression purposes the plasmid with the codon sequence of ClcF or its mutants was transformed in E. coli BL21 (DE3)-CP-RIL.
- For large scale protein production an overnight culture was setup with a colony from the transformation plate. The culture was incubated at 37 °C and 250 rpm for 16 h in LB medium.
- Afterwards 1 L LB medium was inoculated with the overnight culture in a ratio of 1/100 and further incubated at 37 °C and 250 rpm.
- If the culture reached an optical density (OD600 nm) of 0.6 (app. 2-2.5 h) the protein production was induced by adding 1 M IPTG stock solution to a final concentration of 1 mM. The culture was further incubated for 4 h at 30 °C.
- After 4 h induction the cells were harvested at 4,500 x g for 25 min at 4 °C.
- The resulting pellet was resuspended in app. 20 ml wash buffer and again pelleted at 4,500 x g for 45 min at 4 °C.
- The resulting pellet was stored at -20 °C until further usage.
- The mutagenesis-PCR was setup as followed:
- Purification
To get protein of sufficient puritiy ≤ 95 % (estimated from a SDS-gel) a three step strategy including a heat precipitation followed by a two step column chromatography was necessary.
- The cell pellet from 1 liter culture volume was resuspended in 25 ml lysis buffer. A 2 M MgCl2 stock solution was added to a final concentration of 3 mM and 0.5 U DNase were used to the digest DNA.
- Cells were disrupted using a precooled French Press at 1,500 Psi. The cells were passed three times through the French Press to ensure complete lysis.
- The cell debris was removed by centrifugation at 50,000 x g for 1 h.
- The supernatant was heated up to 65 °C for 10 min in a water bath and afterwards cooled down on ice.
- The denatured protein was removed by centrifugation at 50,000 x g for 1 h.
- The resulting supernatant (app. 25 ml) was applied on a Q-sepharose, preequilibrated with IEX-A buffer.
- The protein was applied on the column and the ClcF containing flowthrough (app. 30 ml) was collected.
- Solid ammonium sulfate (AMS) was added to a final concentration of 1.6 M to the flowthrough of the previous step (app. 6 g).
- The resulting solution was applied on a Phenyl-sepharose preequilibrated with HIC buffer. The protein was eluted with a linear gradient to 0 M AMS over 20 column volumes by means of an Aekta chromatography system. ClcF containing fractions were identified by activity assay as described in the underlying paper (Roth et al., 2013).
- ClcF containing fractions were pooled and dialyzed extensively against 250 times the protein volume of 25 mM Tris (pH 7.5) at 4 °C. The dialysis buffer is changed once after 12 h.
- Prior crystallization, the protein was concentrated to 10 mg/ml (determined using the Bradford assay) with a VivaSpin concentrator with a MWCO of 30,000 kDa at 4,000 x g. Aliquots of the protein were directly frozen at -80 °C till further usage.
- The cell pellet from 1 liter culture volume was resuspended in 25 ml lysis buffer. A 2 M MgCl2 stock solution was added to a final concentration of 3 mM and 0.5 U DNase were used to the digest DNA.
- Crystallization
- Based on the recipes of the commercial or published screens (e.g. sparse matrix, grid screen), all necessary buffer solution were prepared as stock solutions at a concentration of 1 M at the relevant pH [Jancarik and Kim, 1991; Brzozowski and Walton, 2001; Hampton Research (see Reference 5); Jena Bioscience (see Reference 6)].
- All salt solutions and precipitant solutions were prepared as stock solutions at the highest concentration possible and filtered if applicable. For ClcF, 1 M Bis-Tris solutions with a pH 6.0 and 7.0 with a spacing of 0.2 pH units are produced (pH adjusted with NaOH/HCl). Furthermore a 2 M MgCl2-solution and a solution of 50 % (w/v) PEG 3350 are necessary. All solutions are prepared with double distilled water.
- The conditions of the respective screens were generated by mixing the appropriate stock solutions, previously prepared, in an Eppendorf tube.
- A screen was setup by a pipetting robot with a reservoir of 90 µl in a Greiner Crystalquick 96 well crystallization plate.
- Drops were setup by pipetting 0.2 µl reservoir solution and 0.2 µl protein solution respectively together. No further mixing is required. The plates are stored afterwards at 19 °C. The plates were then regularly examined (in the first week every day, afterwards weekly) for crystal formation. Crystals usually appears between one day and up to 3 months, but occasionally even longer periods might be necessary to obtain crystals. If a promising condition (crystals, microcrystals or crystalline precipitate) was identified a fine screen was setup in a hanging drop 24 well format usually by varying the pH around the initial condition in a range of ± 1.5 pH units and the precipitant concentration within an applicable range (Figure 1).
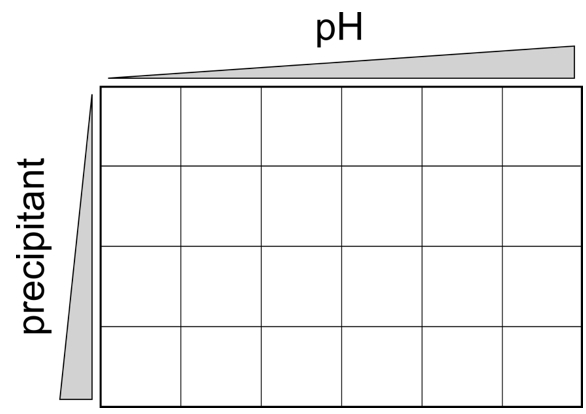
Figure 1. Typical scheme for a 2-dimensional 24 well grid screen, varying pH and precipitant concentration at the same time
- The reservoir was prepared as described and drops were setup with a total drop volume of 2 µl by mixing 1 µl protein with 1 µl reservoir over a reservoir of 500 µl in a 24 well Linbro crystallization plate.
- For orthorhombic crystals of ClcF the screen, with a constant concentration of 0.4 M MgCl2 in all conditions, is shown in Figure 2.
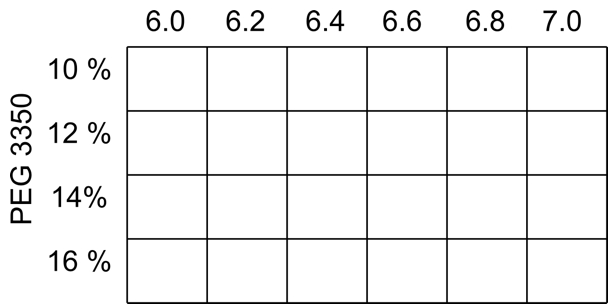
Figure 2. Typical screen for orthorhombic ClcF crystals, varying pH and precipitant concentration at the same time. In all conditions is MgCl2 in a final concentration of 0.4 M.
- For a complex the protein was premixed with 10 mM substrate (5-chloromuconolactone), dissolved in a concentration of 1 M in water at pH 7 (adjusted with NaHCO3). No elongated incubation period is required prior crystallization. The setup of the crystallization plate was carried out as described.

Figure 3. Promising crystalline material within protein precipitate as starting point for further fine screens (left) and typical single crystals of ClcF (right). Further pictures could be found on the Hampton Research website (see Reference 5).
- Based on the recipes of the commercial or published screens (e.g. sparse matrix, grid screen), all necessary buffer solution were prepared as stock solutions at a concentration of 1 M at the relevant pH [Jancarik and Kim, 1991; Brzozowski and Walton, 2001; Hampton Research (see Reference 5); Jena Bioscience (see Reference 6)].
- Cryoprotection
- A condition with good looking crystals was chosen and the reservoir solution was prepared with 2% elevated precipitant concentration supplemented with 25% ethylene glycol.
- Subsequently small amounts, 0.1 to 0.2 µl of the cryosolution was added to the drop and the drop solution slowly exchanged against the cryosolution.
- In the case of the complex the cryosolution contained the precipitant in a final concentration of 25% plus the ligand in a concentration of 10 mM. No additional ethylene glycol is required.
- Finally the crystals were fished with a nylon loop and plunged into liquid nitrogen.
- A condition with good looking crystals was chosen and the reservoir solution was prepared with 2% elevated precipitant concentration supplemented with 25% ethylene glycol.
Recipes
- Lysis buffer/Ion exchange (IEX) buffer A/Hydrophobic interaction chromatography (HIC) buffer A
25 mM Tris-HCl (pH 7.5)
- HIC High salt buffer
25 mM Tris-HCl (pH 7.5) (final pH after all ingredients are added)
1.6 M ammonium sulphate
- Wash buffer
50 mM Tris (pH 7.0)
150 mM NaCl
- Crystallization buffers
pH adjusted with HCl or NaOH
Acknowledgments
The authors used the following standard protocols developed by the mentioned researchers. The Bradford Assay as described in Bradford (1976). The SDS-gel electrophoresis as described in Laemmli (1970). Standard recipes for media and standard buffers are adapted from: “Molecular Cloning.” Green, M. R. and Sambrook, J. Cold Spring Harbor Laboratory Press.
References
- Brzozowski, A. M. and Walton, J. (2001). Clear strategy screens for macromolecular crystallization. J Appl Crystallogr 34(2): 97-101.
- Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
- Hampton Research. www.hamptonresearch.com
- Jancarik, J. and Kim, S. H. (1991). Sparse matrix sampling: a screening method for crystallization of proteins. J Appl Crystallogr 24(4): 409-411.
- Jena Bioscience. www.jenabioscience.com
- Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259): 680-685.
- Roth, C., Gröning, J. A., Kaschabek, S. R., Schlömann, M. and Sträter, N. (2013). Crystal structure and catalytic mechanism of chloromuconolactone dehalogenase ClcF from Rhodococcus opacus 1CP. Mol Microbiol 88(2): 254-267.
- Weiner, M. P., Costa, G. L., Schoettlin, W., Cline, J., Mathur, E. and Bauer, J. C. (1994). Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene 151(1-2): 119-123.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Roth, C., Gröning, J. A. D., Kaschabek, S. R., Schlömann, M. and Sträter, N. (2014). Purification and Crystallization of Chloromuconolactone Dehalogenase ClcF from Rhodococcus opacus 1CP. Bio-protocol 4(8): e1107. DOI: 10.21769/BioProtoc.1107.
Category
Microbiology > Microbial biochemistry > Protein > Isolation and purification
Biochemistry > Protein > Structure
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.