- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Individual-nucleotide-resolution UV Cross-linking and Immunoprecipitation (iCLIP) of UPF1


Published: Vol 4, Iss 7, Apr 5, 2014 DOI: 10.21769/BioProtoc.1085 Views: 15262
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2013
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The fate of mRNA, in particular its stability, localization and rate of translation is regulated by RNA binding proteins assembling to messenger ribonucleoprotein (mRNP) complexes. To investigate the transcriptome-wide RNA binding sites of UPF1, the core factor of nonsense-mediated mRNA decay (NMD), we performed individual-nucleotide-resolution UV cross-linking and immunoprecipitation (iCLIP) (Zund et al., 2013) followed by high-throughput sequencing. The presented protocol is optimized to investigate the RNA-binding sites of UPF1 and is based on previously described studies (Konig et al., 2010; Konig et al., 2011; Hafner et al., 2010). We want to thank the Group of Mihaela Zavolan (Swiss Institute of Bioinformatics, Basel, Switzerland) and Jernej Ule (Medical Research Council Laboratory of Molecualar Biology, Cambridge, UK) for technical support in setting up these experiments.
Materials and Reagents
- Hela cells
- DMEM (powder, high glucose) (Life Technologies, catalog number: 52100-039 )
- Fetal Calf Serum (FCS) (BioConcept, Amimed, catalog number: 2-01F30-I )
- Penicillin-Streptomycin solution (P/S) (1 unit/ml) (BioConcept, Amimed, catalog number: 4-01F00-H )
- Ice
- Trypsin-EDTA (T/E) (BioConcept, Amimed, catalog number: 5-5iF00-H )
- Liquid nitrogen
- DEPC BioChemica (AppliChem GmbH, catalog number: A0881, 0250 )
- 100x HaltTM Protease Inhibitor Cocktail (Thermo Fisher Scientific, catalog number: 1861279 )
- RNase I (cloned) (100 U/µl) (Life Technologies, Ambion®, catalog number: AM2294 )
- NaCl
- Turbo DNase (Life Technologies, Ambion®, catalog number: AM2238 )
- Goat anti-RENT1 antibody (Bethyl Laboratories, catalog number: A300-038A ) (UPF1 is also named RENT1.)
- Phenol
- Chloroform
- Isoamyl alcohol
- Dynabeads® Protein G (Life Technologies, catalog number: 10004D )
- Alkaline Phosphatase Calf Intestinal (CIP) (New England Biolabs, catalog number: M0290S )
- T4 RNA Ligase (10 U/µl) (Thermo Fisher Scientific, catalog number: EL0021 )
- RNasin (Dundee Cell Products, catalog number: RS3100 )
- 3’ RNA linker (5'-P-UGAGAUCGGAAGAGCGGUUCAG-Puromycin-3') (Microsynth AG; http://www.microsynth.ch/)
Note: The RNA is delivered as 5 nmol aliquots, stored at -80 °C and dissolved in 25 µl MQ-water before usage.
- γ-P32-ATP (10 µCi/µl) (HARTMANN ANALYTIC GmbH, catalog number: SCP-401 )
- T4 Polynucleotide Kinase (10 U/µl) (Thermo Fisher Scientific, catalog number: EK0031 )
- 4x NuPAGE® LDS Sample Buffer (Life Technologies, catalog number: NP0007 )
- 20x NuPAGE® MOPS SDS Running Buffer (Life Technologies, catalog number: NP0001 )
- Prestained Protein Ladder (broad range) (New England Biolabs, catalog number: P7710S )
- GlycoBlueTM Coprecipitant (15 mg/ml) (Life Technologies, catalog number: AM9515 )
- Proteinase K (recombinat PCR grade) (Roche Diagnostics, catalog number: 03 115 887 001 )
- RT-primer (Microsynth AG)
Oligo sequence: 5'-P-NNNNAACAGATCGGAAGAGCGTCGTGGATCCTGAACCGC-3'
- dNTP mix (10 mM each) (Thermo Fisher Scientific, catalog number: R0192 )
- AffinityScriptTM Multi-Temp RT (Agilent, catalog number: 600109-51 )
- 40% Acrylamide-Solution (mix 19:1, molecular biology grade) (AppliChem GmbH, catalog number: A3658 )
- Tetramethylethylendiamin (TEMED) (Sigma-Aldrich, catalog number: T22500-100ML )
- Ammonium persulfate (APS) (Sigma-Aldrich, catalog number: A3678-100G )
- PlusOne Repel-Silane ES (GE Healthcare, catalog number: 17-1332-01 )
- Low Molecular Weight DNA Ladder (New England Biolabs, catalog number: N3233L )
- SYBR® Gold Nucleic Acid Gel Stain (Life Technologies, catalog number: S11494 )
- CircLigaseTM II ssDNA Ligase (EpiCentre, catalog number: CL9025K )
- Cut-Oligo (5'- GTTCAGGATCCACGACGCTCTTCAAAA-3') (Microsynth AG)
- BamHI (New England Biolabs, catalog number: R0136S )
- P5_Solexa primer (Microsynth AG)
Oligo sequence: 5'-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3'
- P3_Solexa primer (Microsynth AG)
Oligo sequence: 5'-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3'
- Maximo Taq DNA 2X-Mastermix (GeneOn, catalog number: S114 )
- DMEM-/- (per 900 ml) (see Recipes)
- DMEM+/+ (per 500 ml) (see Recipes)
- Phosphate-bufferd saline (PBS) (pH 7.4) (see Recipes)
- Hypotonic gentle lysis buffer (pH 7.5) (RNase-free) (see Recipes)
- IP-wash buffer (RNase-free) (see Recipes)
- High salt wash buffer (pH 7.5) (RNase-free) (see Recipes)
- PNK buffer (pH 7.5) (RNase-free) (see Recipes)
- PNK buffer w/o DTT (pH 7.5) (RNase-free) (see Recipes)
- Proteinase K buffer (pH 7.5) (RNase-free) (see Recipes)
- TE buffer (pH 7.5) (RNase-free) (see Recipes)
- 10x Tris-borate buffer (TBE) (pH 8.0) (RNase-free) (see Recipes)
- Acidic phenol (see Recipes)
- Solution A (RNase-free) (see Recipes)
- Solution B (RNase-free) (see Recipes)
- Loading buffer for PAA-urea gel (pH 8.0) (RNase-free) (see Recipes)
- 10x DNA loading buffer (see Recipes)
- DEPC treated water/buffer (see Recipes)
- 3 M NaOAc (pH 4.6 and 5.5) (RNAse-free) (see Recipes)
- RNA ligation mix (see Recipes)
- Kinase mix (see Recipes)
- CircLigation mix (see Recipes)
- Oligo annealing mix (see Recipes)
- PCR master mix (see Recipes)
Equipment
- Pure water system: PURELAB Priama (Prima 7) and PURELABULTRA (Ultra Genetic) (ELGA LabWater)
Note: In this protocol pure water from the ELGA system is referred to as MQ-water.
- CO2 incubator (BINDER GmbH, model: 9140-0047 )
- Clear-viewTM Snap-Cap microtubes (1.5 ml, natural, low retention) (Sigma-Aldrich, catalog number: T4816-250EA )
- Multiply®-Pro 0.2 ml Biosphere® (Sarstedt AG, model: 72.727 )
- Filter tips (10 µl, 20 µl and 200 µl) (Axon Lab AG, catalog number: AL60X10 , AL60X20 , AL60X200 )
- Filter tips (1,250 µl) (Greiner Bio-One GmbH, catalog number: 7.750.261 )
- 15 cm tissue culture dishes (TPP Techno Plastic Products, catalog number: 93150 )
- Stratalinker 2400 (254 nm) (Stratagene®, catalog number: 400076 )
- PCR tube
- Cell lifter (TPP Techno Plastic Products, model: 99010 )
- Saran wrap
- Scotch
- GP Millipore Express® PLUS membrane (0.22 µm) (500 ml Funnel, 45 mm Neck Size) (EMD Millipore, catalog number: SCGPT05RE )
- DynaMagTM-2 magnet (Life Technologies, catalog number: 12321D )
- Water bath (temperature adjustable)
- Eppendorf centrifuge 5415R with rotor F45-24-11 (Eppendorf, model: 022621459 and 022636502 )
- CriterionTM Cell (Bio-Rad Laboratories, catalog number: 165-6001 )
- NuPAGE® Novex® 4-12% Bis-Tris Midi Gels (1.0 mmx, 12 + 2 well) (Life Technologies, catalog number: WG1401BOX )
- Midi Gel Adaptor (Life Technologies, model: WA0999 )
- iBlot® Gel Transfer Devise (EU) (Life Technologies, model: IB1001EU )
- iBlot® Transfer Stack (nitrocellulose, regular size) (Life Technologies, catalog number: IB3010-01 )
- 10 ml serological pipette
- POWER PAC 3000 (Bio-Rad Laboratories)
- Lab cycler gradient equipped with Thermoblock 96 (SensoQuest GmbH, models: 011-101 and 012-103 )
- Blotting paper (Genentech, catalog number: BP 002 46579 )
- FUJI screen
- FUJIFILM FLA-3000 phosphorimager
- Costar Spin-X® centrifugation column (Corning, model: 8161 )
- Vertical PAA-gel running apparatus for 180 x 120 mm gels with 1 mm thick spacers and combs (home made)
- Paper clips foldback (51 mm) (Lyreco, catalog number: 161.791 )
- Aluminium plate (90 x 175 mm, thickness 3 mm)
- UV desk (365 nm) (home made)
- 1 ml syringe (BD, PlastipakTM, catalog number: 300013 )
Procedure
- Cell culture and UV cross-linking
- Hela cells are cultivated in Dulbecco’s Modified Eagle Media (DMEM) supplemented with 100 U/ml Penicillin, 100 µg/ml Streptomycin and 10% foetal calf serum in ten 15 cm diameter dishes until they are 80% confluent.
- The media is discarded and cells are covered with 12 ml ice-cold PBS buffer.
- The cells are subjected to 150 mJ/cm2 UV-C light. During the irradiation the dishes are placed on ice.
- After irradiation the cells are covered with 10 ml fresh ice-cold PBS and scraped off using a cell lifter.
- The cells are sedimented in two 50 ml falcon tubes by centrifugation (4 °C, 200 x g, 5 min), shock frozen in liquid nitrogen and stored until usage at -80 °C.
- Hela cells are cultivated in Dulbecco’s Modified Eagle Media (DMEM) supplemented with 100 U/ml Penicillin, 100 µg/ml Streptomycin and 10% foetal calf serum in ten 15 cm diameter dishes until they are 80% confluent.
- Limited RNase I digestion and UPF1 immunoprecipitation
- The cells are thawed on ice and lysed in 7 ml hypotonic gentle lysis buffer for 20 min. The cell debris is removed by centrifugation (4 °C, 13,000 x g, 15 min). The supernatant is recovered and the NaCl concentration is adjusted to 150 mM by the addition of 350 µl of 3 M NaCl.
- For partial RNase digestion, RNase I is diluted 1:1,000 in hypotonic gentle lysis buffer and the cell lysate is treated with 70 µl of the RNase I dilution and 35 µl Turbo DNase with slight shaking in a 37 °C water bath for 3 min.
Note: For each RNA binding protein subjected to iCLIP the RNase I digestion step has to be adjusted de novo in order to obtain 80-200 nt long RNA pieces bound to the protein of interest, suitable for cDNA library preparation and subsequent high-throughput sequencing (see Notes).
- The RNase I treated lysate is rested on ice for 3 min.
- 28 µl of goat anti-RENT1 antibody is added and rotated head over tail at 4 °C for 1 h.
- 240 µl of Dynabeads® Protein G are washed twice with 1 ml PBS buffer and once with 1 ml hypotonic gently lysis buffer. To collect the magnetic Dynabeads® the DynaMagTM-2 Magnet rack is used.
- After 1 h pre-incubation of the lysate with UPF1/RENT1 antibody the washed beads are added and incubated for an additional hour at 4 °C.
- Remove the cell lysate and wash the beads three times with IP wash buffer.
- The cells are thawed on ice and lysed in 7 ml hypotonic gentle lysis buffer for 20 min. The cell debris is removed by centrifugation (4 °C, 13,000 x g, 15 min). The supernatant is recovered and the NaCl concentration is adjusted to 150 mM by the addition of 350 µl of 3 M NaCl.
- Dephosphorylation of the RNA followed by the ligation of the 3’ RNA-linker
- To dephosporylate the RNA pieces covalently bound to UPF1, the Dynabeads® are resuspended in 60 µl dephosporylation mix [55.25 µl MQ-water, 6.5 µl 10x NEBuffer 3, 3.25 µl Alkaline Phosphatase Calf Intestinal (CIP)] and incubated at 37 °C for 20 min.
- The beads are washed twice with 1 ml high salt wash buffer for stringent washing and twice with 1 ml PNK buffer.
- After the last wash step the supernatant is carefully removed and the beads are incubated in 50 µl of RNA ligation mix. The over night ligation of the 3’ RNA linker is performed in a 0.2 ml PCR tube placed at 16 °C in a PCR machine. The lid is kept at constant 50 °C to prevent condensation of the sample.
- To dephosporylate the RNA pieces covalently bound to UPF1, the Dynabeads® are resuspended in 60 µl dephosporylation mix [55.25 µl MQ-water, 6.5 µl 10x NEBuffer 3, 3.25 µl Alkaline Phosphatase Calf Intestinal (CIP)] and incubated at 37 °C for 20 min.
- Radiolabeling of the RNA and isolation of the RNA-UPF1 complex
- The samples is washed twice with 1 ml PNK buffer and the RNA is radioactively labelled by incubating with 60 µl of kinase mix at 37 °C, shaking at 800 rmp for 45 min.
- The radiolabeled sample bound to the beads is washed four times with PNK buffer w/o DTT (The radioactive buffer after the second wash step is kept and used for the hot positioning marks in step 4i.).
- The protein-RNA complexes are eluted from the beads by cooking at 90 °C for 5 min in 45 µl 1x NuPAGE® LDS Sample Buffer.
- The gel running-apparatus is assembled using a BioRAD criterion chamber, a midi gel adaptor and a pre-casted NuPAGE® Novex® 4-12% Bis-Tris midi gel. The running chamber is filled with 500 ml 1x NuPAGE® MOPS SDS Running Buffer.
- The eluted protein-RNA sample (40 µl) is loaded on the NuPAGE® Novex Gel next to 6 µl prestained protein ladder and separated at 200 V for 45-60 min until the bromophenol blue running front is about to leave the gel.
- Disassemble the gel device and cut off the running front marked by the bromophenol blue band. This lower part of the gel contains the non-incorporated γ-P32-ATP and is disposed of as solid radioactive waste.
- The protein-RNA adducts are transferred to nitrocellulose membrane using the iBlot system (for detailed instruction consult the tutorials provided by the manufacturer).
- After the 7 min transfer procedure the transfer stacks are disassembled and disposed of as solid radioactive waste.
- The nitrocellulose membrane is wrapped in saran wrap and three positioning marks are attached at the edge of the membrane [1 µl of the hot PNK buffer put aside in step 4b is spread on a small blotting paper strip (1.5 x 8 mm) and fixed on the saran wrap with a small piece of Scotch. These marks are important to properly align the autoradiograph in step 4k to excise and isolate the UPF1-mRNA complex from the nitrocellulose membrane.].
- For the detection of the P32-labelled RNA-UPF1 complex on the nitrocellulose membrane a FUJI screen is exposed for 1 h and analysed on a FUJIFILM FLA-3000 phosphorimager.
- The autoradiograph of the membrane is printed in real seize format and aligned with the nitrocellulose membrane with help of the positioning markers. The section of the nitrocellulose membrane containing the RNA-UPF1 complex is excised and ground into small pieces using a 1,250 µl pipette tip.
- The RNA is isolated by incubating the membrane pieces with 200 µl proteinase K buffer supplemented with 20% (v/v) proteinase K at 50 °C for 1h.
- The elution is repeated to maximise the elution efficiency.
- The pooled elution fractions (final volume 400 µl) are filtered (spun at 2,000 x g for 2 min) using a costar Spin-X® centrifugation column.
- The samples is washed twice with 1 ml PNK buffer and the RNA is radioactively labelled by incubating with 60 µl of kinase mix at 37 °C, shaking at 800 rmp for 45 min.
- Phenol/chloroform extraction and RNA precipitation
- Add one volume of acidic phenol:chloroform:isoamylalcohol (25:24:1) to the filtered RNA sample and shake at 1,000 rpm (not vortex) for 5 min.
- Spin the sample (16,000 x g, 4 °C, 10 min) and transfer the aqueous phase (350 µl) into a fresh tube.
- Add 35 µl 3 M NaOAc (pH 4.6), 2 µl GlycoBlue Coprecipitant, 400 µl isopropanol; mix and precipitated the RNA over night at -20 °C.
- The RNA is precipitated by centrifugation (16,000 x g, 4 °C, 15 min), washed with 1 ml 70% EtOH and dissolved in 5.5 µl MQ-water.
- Add one volume of acidic phenol:chloroform:isoamylalcohol (25:24:1) to the filtered RNA sample and shake at 1,000 rpm (not vortex) for 5 min.
- Reverse transcription
- Transfer the sample into a 0.2 ml PCR tube.
- 0.5 µl RNasin, 0.5 µl of dNTPs (10 mM each) and 0.5 µl RT-primer (0.5 pmol/µl) are added to the RNA and heated to 70 °C for 5 min (in a PCR thermo cycler).
- After slowly cooling the sample to 25 °C (temperature ramp: 1.8 °C/min), 3 µl of the following RT mix (1 µl 10x AffinityScript RT buffer, 1 µl 0.1 M DTT, 1 µl Affinity Script Multi-Temp RT) is added.
- Continue the reverse transcription with the following thermal program:
25 °C
15 min
50 °C
60 min
4 °C
hold
- The cDNA is transferred to 1.5 ml test tube, precipitated by adding 90 µl TE buffer, 0.5 µl GlycoBlue Coprecipitant, 10 µl NaOAc (pH 5.5), 250 µl 100% EtOH and a subsequent incubation at -20 °C for over night.
- The cDNA is pelleted by centrifugation (4 °C, 16,000 x g, 15 min).
- The pellet is washed with 70% EtOH.
- Resuspend the cDNA in 10 µl loading buffer for PAA-urea gels and cook at 70 °C for 5 min.
- Mix 2 µl low molecular weight marker with 2 µl 6x loading buffer and 8 µl loading buffer for PAA-urea gels (do not cook the marker).
- Transfer the sample into a 0.2 ml PCR tube.
- Cast 6% TBE-urea gel
- Wash and dry the glass plates (180 x 120 mm) carefully.
- Apply PlusOne repel-silane ES with a soft paper towel to the inner surface of the glass plates.
- Assemble the glass plates and the spacers (1 mm thickness) and fix the setup with two file clips centred on the spacers. Lay the assembled glass plates on an empty tip box. Casting the gel in horizontal position makes sealing of the boarders redundant.
- Prepare 25 ml 6% gel solution by mixing 7.5 ml solution A with 17.5 solution B. Start the polyimerization by adding 125 µl 10% APS and 12.5 µl TEMED.
- The gel solution is applied with a 10 ml serological pipette to edge of the glass plate on the side where the comb will be inserted. Surface tension and slight tapping on the glass plate ensures an even and bubble-free distribution of the gel solution.
- Introduce the comb and fix the glass plates with additional file clips.
- Polymerize the gel for 1 h.
- Wash and dry the glass plates (180 x 120 mm) carefully.
- Run 6% TBE-urea gel
- Clamp the gel assembled with an aluminium plate into the gel running chamber (The 90 x 175 x 3 mm aluminium plate ensures even heat distribution and therewith even running of the samples in the gel.) and fill the apparatus with 1x TBE buffer.
- Wash the slots carefully with a syringe.
- Pre-run the gel (10 W, 30 min).
- Wash the slots again.
- Load the low molecular weight DNA ladder and the cDNA sample leaving 2-3 empty lanes in between.
- Run the gel (5 W, 70 min) until the bromophenol blue band is 2 cm above the bottom of the gel.
- Clamp the gel assembled with an aluminium plate into the gel running chamber (The 90 x 175 x 3 mm aluminium plate ensures even heat distribution and therewith even running of the samples in the gel.) and fill the apparatus with 1x TBE buffer.
- Isolate cDNA
- Cut the gel in order to separate the lane containing the size ladder from the lane containing the sample.
- Stain the marker-containing gel part with 1x SYBR-gold in 1x TBE buffer (for 15 min) and wrap the part of the gel containing the sample in saran wrap.
- Visualize the DNA ladder on a 365 nm UV-desk and copy the position of the size ladder on a transparency.
- With help of the transparency showing the size ladder (placed next to the unstained gel part) excise the portion of the gel containing the cDNA covering a size range form 150-300 nt.
- Crush the gel piece with a syringe plunger (from a 1 ml syringe) and extract the cDNA with 400 µl TE buffer (shaking at 2,000 rpm, 37 °C, 2 h).
- Filter the supernatant with a Costar Spin-X® centrifugation column (2 min, 2,000 x g).
- Precipitate the cDNA by adding 1 µl GlycoBlue Coprecipitant, 40 µl NaOAc (pH 5.5) and 1 ml 100% EtOH and incubate at -20 °C for 1 h.
- Pellet the cDNA by centrifugation (4 °C, 16,000 x g, 15 min).
- Wash the pellet with 1 ml 70% EtOH.
- Dissolve the cDNA in 6.7 µl MQ-water.
- Cut the gel in order to separate the lane containing the size ladder from the lane containing the sample.
- Introduction of the 5’ primer to the cDNA by circularization and subsequent linearization
- To perform the circularization add 1.5 µl CircLigation mix to the 6.7 µl cDNA followed by the incubation in a PCR thermo cycler at 60 °C for 1 h.
- To linearize the single stranded circular cDNA, a DNA cut-oligo is annealed to a region in the RT-primer generating a double-stranded DNA stretch harbouring a BamHI site.
Prepare the oligo annealing mix and add 30 µl of it to the circular cDNA. The sample is heated to 90 °C and slowly cooled down to 25 °C (in PCR thermo cycler, 1 °C/min temperature ramp).
- To cleave the circular cDNA, 2 µl BamHI restriction enzyme is added and incubated at 37 °C for 45 min.
- Precipitate the linearized cDNA by adding 50 µl TE buffer, 0.5 µl GlycoBlue Coprecipitant, 10 µl NaOAc (pH 5.5) and 250 µl 100% EtOH prior to the incubation at -20 °C for over night.
- Pellet the linearized cDNA by centrifugation (4 °C, 13,000 x g, 15 min).
- Wash the pellet with 70% EtOH.
- The precipitated cDNA is dissolved in 50 µl MQ-water.
- To perform the circularization add 1.5 µl CircLigation mix to the 6.7 µl cDNA followed by the incubation in a PCR thermo cycler at 60 °C for 1 h.
- PCR amplification of the cDNA
The adapters needed for high-throughput Illumina sequencing are introduced by PCR using the P5_Solexa and the P3_Solexa primers.- Pilot PCR
In order to determine the number of cycles needed for a proper amplification of the cDNA library, a 100 µl test PCR is performed. To set up the test PCR, combine 10 µl cDNA, 40 µl PCR master mix and 50 µl Maximo Taq DNA mastermix and run the following PCR program:
95 °C for 5 min
34 cycles of (95 °C for 45 s, 50 °C for 80 s, 72 °C for 1 min)
To analyze the intermediate PCR products after cycle 20, 22, 24, 26, 28, 30, 32 and 34, a 12 µl sample is taken and mixed with 3 µl 10x DNA loading buffer.
Of each sample separate 7.5 µl on a 2.5% agarose gel to determine the number of cycles (Nthreshold) needed to generate just enough PCR product resulting in a band visible on the agarose gel. Overamplification should be avoided as it drastically reduces the complexity of the sequencing library.
- Preparative PCR
For the preparative PCR amplification of the cDNA library four 100 µl PCR reactions are run with the following cycles:
95 °C for 5 min
Nthreshold cycles of (95 °C for 45 s, 50 °C for 80 s, 72 °C for 1 min)
72 °C for 5 min
- Clean up the preparative PCR product
The four PCR products are combined, mixed with 400 µl phenol:chlorofom:isoamylalcohol (25: 24: 1) and rotated head over tail at room temperature for 5 min.
After centrifugation (4 °C, 13,000 x g, 15 min) recover 350 µl of the aqueous phase, add 35 µl NaOAc (pH 5.5), 1 µl GlycoBlue Coprecipitant and 800 µl 100% EtOH and precipitate over night at -20 °C.
Precipitate the cDNA library by centrifugation (4 °C, 13,000 x g, 15 min), discard the supernatant and wash the pellet with 1 ml 70% EtOH.
Dissolve the DNA in 20 µl TE buffer, add 3 µl DNA loading buffer and separate the cDNA on a 2.5% agarose gel. The DNA band is excised and isolated using the Wizard SV Gel PCR Clean-Up System.
Note: Do not denature the DNA, dissolve the gel at 50 °C as shortly as possible!
- Pilot PCR
- Submit your cDNA library for high throughput sequencing on an Illumina HiSeq 2000 sequencer performing single-end reads.
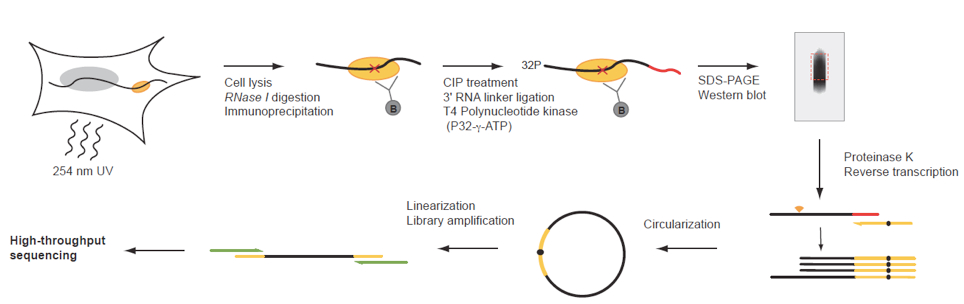
Figure 1. iCLIP procedure of endogenous UPF1. Schematic illustration of the iCLIP procedure. CIP, Alkaline Phosphatase Calf Intestinal. (figure adapted from Reference 1).
Notes
- Throughout the entire iCLIP protocol Clear-viewTM Snap-Cap microtubes are used to minimize the absorption of the sample by the plastic and therewith to prevent sample loss.
- The usage of the NuPAGE® Novex® 4-12% Bis-Tris Midi Gel in combination with the NuPAGE® MOPS SDS Running Buffer keeps a constant pH 7 during the running procedure and therefore prevents the RNA from alkaline hydrolysis.
- To successfully perform iCLIP it is crucial to establish an efficient immunoprecipitation (IP) protocol optimized for your RNA binding protein of interest. Different antibodies and various buffer conditions have to be tested.
- For every RNA-binding protein, different RNase I concentrations have to be tested in the limited RNase digestion step in order to obtain RNA fragments with a suitable size (80-200 nt) for cDNA library preparation and subsequent high-throughput sequencing. Especially, working with RNase I, which has no nucleotide specificity, requires careful titration of the RNase I working concentration in order to prevent over-digestion of the RNA. RNase I dilutions from 1: 50 up to 1: 2,000 appeared to be a suitable range for an initial titration experiment. Harsh RNase digestion results in a clear band of the protein-RNA adduct on the autoradiogram after Western blotting. Reducing the RNase concentration leads to a shift of the signal towards higher molecular weight due to the increased heterogeneity in size of the RNA bound to the protein (Figure 2). An 80 nt long RNA contributes approximately 26 kDa to the protein-RNA complex.
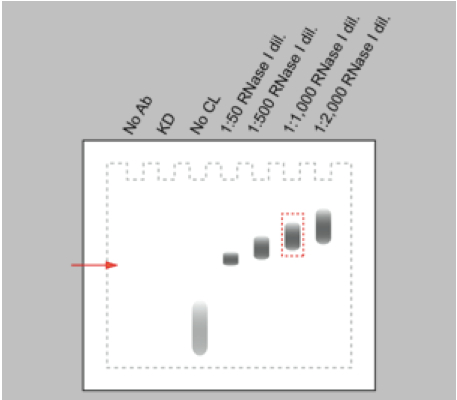
Figure 2. Schematic representation of an autoradiograph of immunopurified and 32P-labelled RNA-protein complexes transferred to nitrocellulose membrane. Immunopurification performed without antibody (no Ab), with cells either depleted of the RNA-binding protein (KD) or not irradiated with UV-C light (no CL) are controls for the specificity of the RNA-protein signal. Increasing dilutions of the RNase I (1:50 to 1:2,000) induce a shift to higher molecular weight of the immunopurified RNA-protein complex. The red arrow marks the electrophoretic mobility of the free RNA-binding protein.
- To control for the IP specificity, knocking down the protein of interest or omitting the antibody during the IP procedure should prevent the accumulation of the protein-RNA complex. In the same way, the absence of a protein-RNA complex on the autoradiogram of the western blot is expected when cells are not cross-linked with UV-light. The free radiolabelled RNAs result in a signal running at low molecular weight (Figure 2).
Recipes
Note: All buffers used in the iCLIP protocol have to be sterile, RNase-free and clean of contaminating nucleic acids. To minimize contaminations, it is recommended to have an extra set of reagents reserved for iCLIP experiments. All buffers are prepared with MQ-water and if possible treated with DEPC. Duran bottles, glassware and spatulas are baked at 180 °C for 2 h. Polycarbonate or polystyrene materials (e.g. magnetic stirrers) are soaked in 3% hydrogen peroxide or 2 M NaOH for 10 min and extensively rinsed with DEPC-treated water before use. If a buffer cannot be DEPC treated (e.g. Tris buffers), prepare the buffer in DEPC or MQ-water and filter it with a 0.22 µm filter.
- DMEM-/- (per 900 ml)
12 g DMEM/F12 powder
2.48 g NaHCO3
Make up to 900 ml with ddH2O
Adjust pH to 7.2 with 32% HCl or 10 M NaOH
Sterilize per filtration with bottle top filter (0.22 µm)
- DMEM+/+ (per 500 ml)
Supply 450 ml DMEM-/- with 50 ml FCS and 5 ml P/S
- Phosphate-bufferd saline (PBS)
137 mM NaCl
10 mM Na2HPO4
2.7 mM KCl
2 mM KH2PO4
Adjust pH to 7.4 with HCl
- Hypotonic gentle lysis buffer (RNase-free)
10 mM Tris-HCl (pH 7.5)
10 mM NaCl
2 mM EDTA
0.5% (v/v) Triton X-100
Prior to usage supplement with 1x HaltTM protease inhibitor cocktail
- IP-wash buffer (RNase-free)
50 mM HEPES-NaOH (pH 7.5)
300 mM KCl
0.05% (v/v) NP-40
0.5 mM DTT
Prior to usage supplement with 1x HaltTM protease inhibitor cocktail
- High salt wash buffer (RNase-free)
50 mM Tris-HCl (pH 7.5)
1 M NaCl
1 mM EDTA
1% (v/v) NP-40
0.1% (w/v) SDS
0.5% (w/v) sodium deoxychlate
Prior to usage supplement with 1x HaltTM protease inhibitor cocktail
- PNK buffer (RNase-free)
50 mM Tris-HCl (pH 7.5)
10 mM MgCl2
5 mM DTT
Prior to usage supplement with 1x HaltTM protease inhibitor cocktail
- PNK buffer w/o DTT (RNase-free)
50 mM Tris-HCl (pH 7.5)
10 mM MgCl2
Prior to usage supplement with 1x HaltTM protease inhibitor cocktail
- Proteinase K buffer (RNase-free)
50 mM Tris-HCl (pH 7.5)
50 mM NaCl
1% (w/v) SDS
10 mM EDTA
- TE buffer (RNase-free)
10 mM Tris-HCl (pH 7.5)
1 mM EDTA
- 10x Tris-borate buffer (TBE) (RNase-free)
890 mM Tris
890 mM Boric acid
20 mM EDTA (pH 8)
- Acidic phenol
Melt phenol at 60 °C
Equilibrate against 2x MQ-water and 0.1% hydroxyquinolone
Aliquot the phenol-water suspension in 50 ml falcon tubes and stored at -20 °C
- Solution A (RNase-free)
20% Polyacrylamide (29:1)
1x TBE
8 M Urea
- Solution B (RNase-free)
1x TBE
8 M Urea
- Loading buffer for PAA-urea gel (RNase-free)
20 mM EDTA (pH 8.0)
91% (v/v) deionized formamide
0.5% (w/v) bromophenol blue
0.5% (w/v) xylene cyanol
- 10x DNA loading buffer
0.6% (w/v) SDS
62.5% (v/v) glycerol
0.5% (w/v) bromophenol blue
- DEPC treated water/buffer
Water/buffer
0.1% (v/v) DEPC
Stir over night
Autoclave twice in order to inactivate DEPC
- 3 M NaOAc (pH 4.6 and 5.5) (RNAse-free)
3 M NaOAc
Adjust pH to 4.6 and 5.5 with 32% HCl
- RNA ligation mix
28 µl MQ-water
5 µl 10x reaction buffer for T4 RNase ligase
5 µl 1 mg/ml BSA
1 µl RNasin
10 µl 20 µM 3’ RNA linker
1 µl 10 U/µl T4 RNA ligase
- Kinase mix
38 µl MQ-water
6 µl 10x reaction buffer A (for forward reaction)
10 µl of 10 µCi/µl γ-P32-ATP
6 µl 10 U/µl T4 Polynucleotide Kinase
- CircLigation mix
0.8 µl 10x CircLigase buffer II
0.4 µl 50 mM MnCl2
0.3 µl CircLigase II
- Oligo annealing mix
21 µl MQ-water
4 µl 1 mg/ml BSA
4 µl Buffer 4
1 µl 10 µM Cut Oligo
- PCR master mix
2.8 µl 100 µM P5_Solexa
2.8 µl 100 µM P3_Solexa
194.5 µl MQ-water
Acknowledgments
We thank R. Bruggmann and S. Neuenschwander (Institute of Cell Biology, University of Bern, Bern, Switzerland) for initial bioinformatics support, and H. le Hir (Institut de Biologie, École Normale Supérieure, Paris, France) for technical hints with the CLIP. This protocol is optimized to investigate the RNA-binding sites of UPF1 and is based on previously described studies (Konig et al., 2010; Konig et al., 2011; Hafner et al., 2010). Further, we want to thank the Group of Mihaela Zavolan (Swiss Institute of Bioinformatics, Basel, Switzerland), Jernej Ule (Medical Research Council Laboratory of Molecualar Biology, Cambridge, UK) for technical support in setting up these experiments, and Andreas R. Gruber (Swiss Institute of Bioinformatics, Basel, Switzerland) for performing the bioinformatics analysis.
References
- Zünd, D., Grüber, A. R., Zavolan, M. and Mühlemann, O. (2013). Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3' UTRs. Nat Struct Mol Biol 20(8): 936-943.
- König, J., Zarnack, K., Rot, G., Curk, T., Kayikci, M., Zupan, B., Turner, D. J., Luscombe, N. M. and Ule, J. (2010). iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17(7): 909-915.
- König, J., Zarnack, K., Rot, G., Curk, T., Kayikci, M., Zupan, B., Turner, D. J., Luscombe, N. M. and Ule, J. (2011). iCLIP-transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. JoVE (50).
- Hafner, M., Landthaler, M., Burger, L., Khorshid, M., Hausser, J., Berninger, P., Rothballer, A., Ascano, M., Jr., Jungkamp, A. C., Munschauer, M., Ulrich, A., Wardle, G. S., Dewell, S., Zavolan, M. and Tuschl, T. (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141(1): 129-141.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zünd, D. and Mühlemann, O. (2014). Individual-nucleotide-resolution UV Cross-linking and Immunoprecipitation (iCLIP) of UPF1. Bio-protocol 4(7): e1085. DOI: 10.21769/BioProtoc.1085.
Category
Biochemistry > RNA > RNA-protein interaction
Molecular Biology > RNA > mRNA translation
Biochemistry > Protein > Immunodetection > Immunoprecipitation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.