- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Retinal Explant Culture

Published: Vol 4, Iss 2, Jan 20, 2014 DOI: 10.21769/BioProtoc.1032 Views: 22402
Reviewed by: Xuecai Ge
Original research article
The authors used this protocol in:

Nov 2012
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
A particularly powerful culture method for the retina is the explant assay, which consists in culturing a small piece of retina on an organotypic filter. Retinal explants can be prepared any time between embryonic day 13 (E13) and postnatal day 4 (P4). Although retinal ganglion cells tend to degenerate shortly after they are generated in explants, and photoreceptor cells do not grow extended outer segments, the explants will develop very similarly to a retina in vivo and generate all the different retinal cell types that will migrate to the appropriate layer. The retinal explant culture assay is particularly useful in cases where a mouse mutant is embryonic lethal and its retinal development cannot be studied in vivo. Because retinal explants can be prepared from embryonic animals and electroporated or infected with viral vectors, it is also a useful approach for the study of gene function at embryonic stages. Here, we present a retinal explant culture method that we have used extensively in various publications (Kechad et al., 2012; Cayouette et al., 2003; Cayouette and Raff, 2003; Elliott et al., 2008).
Keywords: Cell lineageMaterials and Reagents
- Animals
We typically use retinas from albino Sprague Dawley rats or mice. Explants can be prepared from animals aged between embryonic day 12 to post-natal day 4. - DPBS (Life Technologies, catalog number: 14040 )
- DMEM (Life Technologies, catalog number: 10569-010 )
- Fetal bovine serum (FBS) (Wisent, catalog number: 080-450 )
- Penicillin/streptomycin (Life Technologies, catalog number: 15070 )
- 70% ethanol
- Fungizone antimycotic (Life Technologies, catalog number: 15290026 )
- Sucrose (Sigma-Aldrich, catalog number: S0389 )
- O.C.T. compound (Somagen, catalog number: 4583 )
- Explant medium
- 4% Paraformadehyde (PFA) solution (Electron Microscopy Sciences, catalog number: 15710 ) (see Recipes)
- 20% sucrose/OCT (1:1) (see Recipes)
- 20% sucrose solution (see Recipes)
Equipment
- Millicell organotypic insert (EMD Millipore, catalog number: PICMORG50 )
- 35 mm dish or 6 well plates
- 37 °C and 5% CO2 incubator
- 100 mm dish
- 150 mm dish
- Straight forcep
- FIne forcep (Fine Science Tools, catalog number: 11252-23 )
- Curved forcep (Fine Science Tools, catalog number: 91197-00 )
- Sharp scissor
- Sharp scapel (Fine Science Tools, catalog number: 10315-12 )
- Razor blade
- P1000 pipette
- CO2 chamber
- Cryomold 15 x 15 x 5 mm (VWR International, catalog number: CA60872-016 )
Procedure
Day of Experiment
- Prepare the explant medium (see Recipes for the preparation of the stock solutions)
- Explant medium
In a 50 ml tube, add: 45 ml of DMEM
5 ml of fetal bovine serum (FBS)
500 µl of pen/strep 100x
50 µl of Fungizone antimycotic
To equilibrate the medium at the right temperature and pH, put the lid on the 50 ml tube and unscrew ¼ of a turn. Place the tube in the incubator at 37 °C and 5% CO2 until needed.
- Explant medium
- Preparation of the culture plates
- The explants are cultured on a Millicell organotypic insert. We typically use 35 mm dish or 6 well plates to put the insert in.
- Fill the dish (or well) with 1.2 ml of equilibrated explants medium and place the insert gently over it with sterile forceps.
- Place the dish (or plate) in the incubator at 37 °C and 5% CO2 until needed.
- Dissection of the retinas
Note: The dissection should be carried out under sterile conditions. Spray the working area with 70% ethanol and use a flame. Sterilize all the instruments before use.
Prepare two 100 mm dish filled with 10 ml of sterile DPBS, one to transfer the heads and the other to dissect the eyes and two 35 mm dish filled with 2 ml of sterile DPBS to collect the retinas.- Collection of the embryos
- Euthanize the animal in a CO2 chamber.
- Place the animal on its back and clean the abdomen with 70% ethanol.
- Pinch the skin with a straight forcep, lift the skin and muscle and make a V shape opening using scissors on both side of the abdomen, starting from the lower abdomen and cutting all the way up on the side of the animal. Lift the skin and muscle upward to reveal the internal organs and uterus.
- Cut out the uterus and transfer it to a 150 mm dish.
- Remove the embryos from the uterus and decapitate the heads with sharp scissors.
- Transfer the heads to a 100 mm dish filled with DPBS.
- Euthanize the animal in a CO2 chamber.
- Dissection of the retinas
- Carefully remove the skin that covers the eye.
- Place a curved forcep of both side of the eye and apply a gentle pressure. The eye should pup out.
- Pinch the back of the eye and pull.
- Transfer the eye in a 100 mm dish filled with DPBS.
- With the lens facing downward, secure the eye in position with a fine forceps and remove the optic nerve (Figure 1, steps 1-2).
- Introduce the tip of another forceps through the optic nerve head and between the choroid and retina to secure the sclera.
- Now that the sclera is secured, you need to gently tear it open with both forceps from the optic nerve head to the cornea to expose the retina and lens (Figure 1, steps 3-4).
- Carefully detach the lens and place the retinas in a 35 mm dish filled with DPBS (Figure 1, steps 5-6).
- From E18-19 onward, it is required to remove the blood vessels covering the retina to avoid endothelial cell contamination in the culture.
- The blood vessels form a reddish membrane overlying the inner face of the retina. The membrane is easily removed by pinching and pulling with fine forceps.
- Transfer the retinas in a 35 mm dish filled with DPBS.
- With the lens facing downward, secure the eye in position with a fine forceps and remove the optic nerve (Figure 1, steps 1-2).
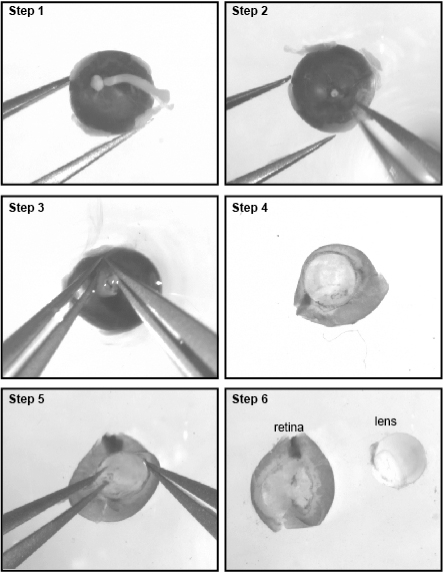
Figure 1. Procedure for retina dissection. Steps 1-5 show the procedure to dissect the retina from the eyeball. Step 6 shows the dissected retina separated from the lens. - Carefully remove the skin that covers the eye.
- Collection of the embryos
- Preparation of explants
- For retinas older than embryonic day 15, cut small pieces of around 5 mm (Figure 2, steps 1-2). For younger retinas, place the whole retina on the insert.
- With a razor blade, cut the edge of a P1000 pipette tip to make a lager opening. Transfer the explants from the 35 mm dish to the insert. It doesn’t matter which side of the explants is facing upward on the insert. Gently lay the explants as flat as possible (Figure 2, step 3). Try not to fold or tear the explants as this could lead to defect in the formation of the layers and make the analysis difficult.
- Place the dish in the incubator at 37 °C and 5% CO2. The culture should be left until the retina is fully developed (around post natal day 14 in vivo). For explants started at embryonic day 13, this would mean 20 days in culture. For explants started at post-natal day 0, this would mean 14 days in culture.
- The medium should be changed every 3 days by replacing 80% of the medium with fresh one.
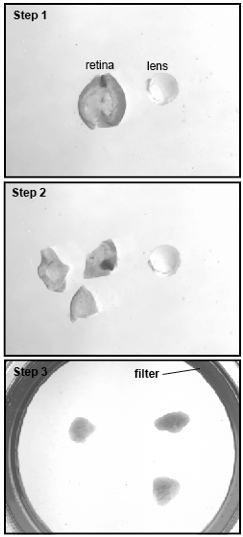
Figure 2. Preparation of the explants. Starting from an entire retina (Step 1), small pieces are cut (Step 2) and placed on the organotypic filter (Step 3).
- For retinas older than embryonic day 15, cut small pieces of around 5 mm (Figure 2, steps 1-2). For younger retinas, place the whole retina on the insert.
- Fixation of the explants
- After the desired culture period, remove the insert from the dish and aspirate the culture medium.
- Replace it with freshly made 4% PFA. Place the insert back in the dish. Gently add 1 ml of 4% PFA on top of the insert. The PFA solution needs to be at room temperature to help maintain the explant morphology.
- Incubate 30 min. Gently rinse 3 times with PBS.
- After the desired culture period, remove the insert from the dish and aspirate the culture medium.
- Preparation of the explants for cryosectionning
- Replace the PBS with a solution of 20% sucrose. Make sure that the explants are well covered. Incubate at 4 °C overnight.
- For cryoprotection, fill a small cryomold with a mixture of 20% sucrose/OCT (1:1).
- Remove the 20% sucrose solution from the top of the insert, take out the insert from the dish and, using a sharp scapel, cut the membrane around each explant.
- Pick the piece of membrane/explant with a fine forceps and place it in the cryomold. Gently push down the explants and lay them at the bottom of the cryomold.
- Make sure it is covered with enough 20% sucrose/OCT solution.
- Snap-freeze in liquid nitrogen (do not immerse the cryomold in liquid nitrogen, just touch the surface of the nitrogen with the bottom of the mold) and store at -80 °C until ready to section.
- The section thickness can vary between 10 and 30 μm depending on the type of analysis needed.
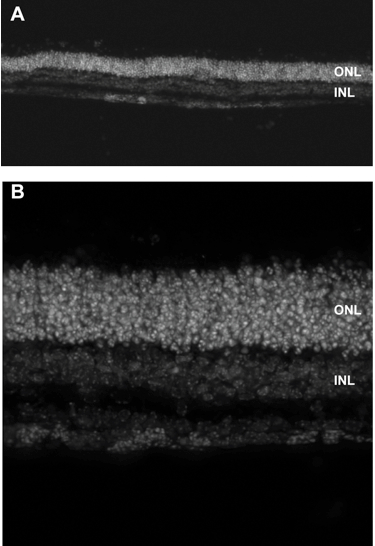
Figure 3. Micrographs of retinal explant cryosections at 10x (A) or 40x magnification after 14 days in culture. The retina was counterstained with Hoechst to visualize the nuclei. The retinal layers are identified, ONL: outer nuclear layer, INL: inner nuclear layer.
- Replace the PBS with a solution of 20% sucrose. Make sure that the explants are well covered. Incubate at 4 °C overnight.
Recipes
- 4% paraformaldehyde
10 ml of 16% paraformaldehyde
26 ml of deionised water
4 ml of 10x PBS (pH 7.4) - 20% sucrose
Dissolve 20 g of sucrose in 80 ml of 1x PBS (pH 7.4)
Once in solution, complete the volume to 100 ml with 1x PBS (pH 7.4) - 20% sucrose/O.C.T. (1:1)
20 ml of 20% sucrose solution
20 ml of O.C.T. compound
Mix thoroughly
Acknowledgments
Funding for this work was provided by the Canadian Institutes of Health Research and the Foundation Fighting Blindness Canada. This protocol was adapted from procedures published in Kechad et al. (2012) and Elliott et al. (2008). The authors wish to thank members of the Cayouette lab, past and present, for continuous support and improvements on the protocol over the years.
References
- Cayouette, M., Barres, B. A. and Raff, M. (2003). Importance of intrinsic mechanisms in cell fate decisions in the developing rat retina. Neuron 40(5): 897-904.
- Cayouette, M. and Raff, M. (2003). The orientation of cell division influences cell-fate choice in the developing mammalian retina. Development 130(11): 2329-2339.
- Elliott, J., Jolicoeur, C., Ramamurthy, V. and Cayouette, M. (2008). Ikaros confers early temporal competence to mouse retinal progenitor cells. Neuron 60(1): 26-39.
- Kechad, A., Jolicoeur, C., Tufford, A., Mattar, P., Chow, R. W., Harris, W. A. and Cayouette, M. (2012). Numb is required for the production of terminal asymmetric cell divisions in the developing mouse retina. J Neurosci 32(48): 17197-17210.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Jolicoeur, C. and Cayouette, M. (2014). Retinal Explant Culture. Bio-protocol 4(2): e1032. DOI: 10.21769/BioProtoc.1032.
- Kechad, A., Jolicoeur, C., Tufford, A., Mattar, P., Chow, R. W., Harris, W. A. and Cayouette, M. (2012). Numb is required for the production of terminal asymmetric cell divisions in the developing mouse retina. J Neurosci 32(48): 17197-17210.
Category
Neuroscience > Development > Retinal culture
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.