摘要:对生物种群进行遗传多样度分析,有助于探究其有效群体大小(Ne)变化以及评估其进化潜力。核苷酸多样度(θπ)反映了群体内不同个体DNA序列间的观察平均碱基差异占比,被广泛用于表征种群的遗传多样度水平。与基于少量DNA片段计算得到的θπ(不同遗传区域重组率和突变速率的差异导致有效群体大小和进化潜力的评估存在偏差)相比,基于全基因组的单核苷酸多态性位点(single nucleotide polymorphism,SNP)计算得到的θπ分布和平均值更能全面地反映目标种群的遗传多样度水平。
关键词: 有效群体大小, SNP, θπ, VCFtools
前言
对生物种群基因组遗传多样度的计算和估计,一方面,可以比较不同群体间有效群体规模的大小,并评估进化潜能。另一方面,可以解析基因组中哪些遗传区域经历了快速演化事件(例如自然选择)。目前,人们以不同参数来表征生物种群的遗传多样度,例如:单倍型多样度、杂合度和核苷酸多样度等。单倍型多样度一般适用于对较小遗传区域的分析;杂合度聚焦于单个个体,但是忽略了对种群所有样本结果的标准化;核苷酸多样度是广义的群体杂合度,被广泛用于表征一个种群的遗传多样度。在此,我们将介绍如何利用群体基因组数据,对目标种群的遗传多样度分布特征进行分析和探究。
软件运行环境及信息
- 本文中所有软件的运行及数据处理和分析均在Linux操作系统环境上进行(相关学习教程请参考:http://linux.vbird.org)
- VCFtools v0.1.16 (https://vcftools.github.io/man_latest.html;Danecek et al.,2011)
- R v4.0.2 (https://cran.r-project.org/src/base/R-4)
实验步骤
- 获取群体基因组SNPs
基于群体的二代基因组重测序数据,运用目前主流的BWA (Version: v0.7.7)+ SAMtools (Version: v1.10) + Picard (Version: v2.18.11) + GATK(Version: v3.7)分析流程获得该群体所有个体的基因组突变位点信息(SNPs)(具体操作流程请参见相关软件的使用说明文档),并生成全基因组VCF文件。本文,我们以网上已发表的川金丝猴群体基因组重测序数据为例(Kuang et al. 2020)进行基因组遗传多样度分布的分析(以核苷酸多样度为表征)。下载的基因组重测序数据经上述分析流程处理后获得全基因组VCF文件(文件格式及具体描述可以参见图1及在线文档https://github.com/samtools/hts-specs)。
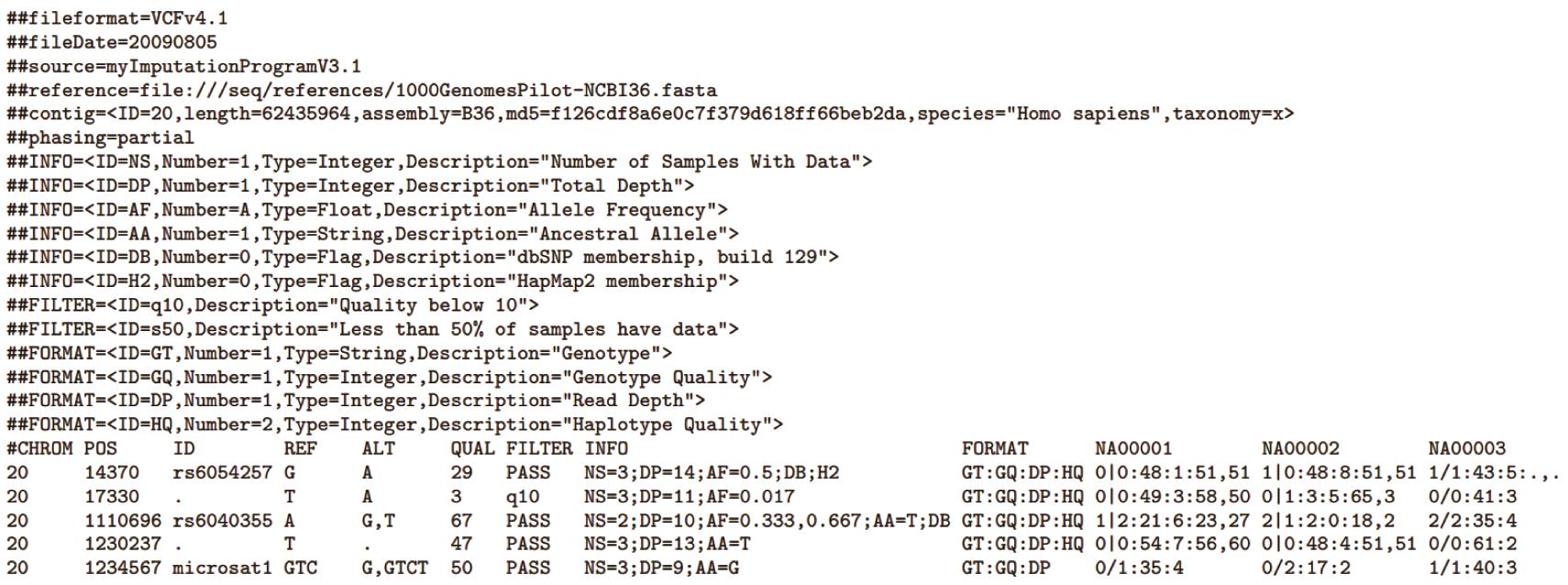
图1. VCF文件的标准格式示例
- 全基因组核苷酸多样度计算
基于单个种群的全基因组VCF文件,可以使用VCFtools对该文件中的所有个体进行扫描以计算该群体的基因组核苷酸多样度。当VCF文件包含来自不同自然种群的个体时需要以--keep(保留一个或多个个体)或--remove(剔除一个或多个个体)选项对样本加以区分和筛选,以此确保分析结果的准确性。对于单个群体的VCF文件,其基因组窗口化的核苷酸多样度计算以如下命令进行:
vcftools --vcf Samples.vcf --window-pi 20000 --out Samples.20K
注:--vcf Samples.vcf 表示输入文件为Samples.vcf;--window-pi 20000表示以20K的窗口大小扫描该VCF文件;--out Samples.20K表示输出结果的文件名前缀。窗口大小的设定一般要考虑以下因素:1.如果在上游SNPs的获取等数据处理中,其所用参考基因组质量不高且序列拼装得较为碎片化时,则建议窗口不要取得太大(容易导致在拼装很短的scaffold上出现扫描区域长度的不均匀),一般选择10或20K大小的窗口进行扫描;2.如果想比较不同种群间基因组水平的遗传多样度分布差异,一般选取小于或等于100K的窗口大小(如:10K,20K或50K等)进行扫描和结果的横向比较即可;3. 如果想通过对某个种群全基因组核苷酸多样度分布的解析来鉴定其基因组中快速进化(自然选择)的遗传区域时,则建议选择10-20K左右的窗口进行扫描(窗口设置太大容易导致选择信号被基因组的背景信号掩盖)。
vcftools --vcf Samples.vcf --window-pi 20000 --window-pi-step 10000 --out Samples.20-10K
注:该命令也是以20K窗口大小扫描整个VCF文件并计算每个窗口下该群体的核苷酸多样度;但该命令指定了相邻两个窗口交叠10K,即窗口以10K的步长在基因组上进行滑动和计算 (加入滑动窗口步长是为了探究连续滑动窗口之间核苷酸多样度是否存在剧烈波动;在对自然选择区域的鉴定分析中有利于较为精细的定位发生自然选择的核心区域。该参数一般以窗口大小的一半进行设定)。
基于上述两条命令,可分别获得Samples.20K.windowed.pi与Samples.20-10K.windowed.pi 结果文件(其文件格式如下图2,3所示)。
Samples.20K.windowed.pi:
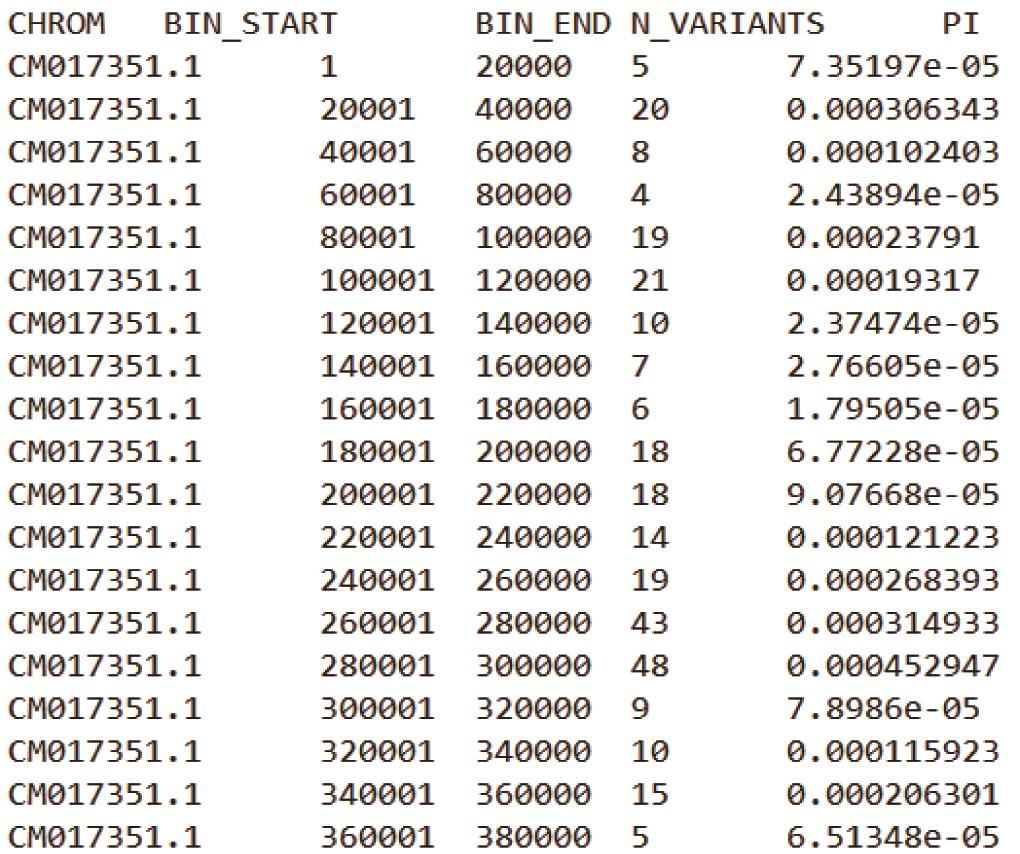
图2. 无步长20K滑动窗口的θπ结果文件
Samples.20-10K.windowed.pi:
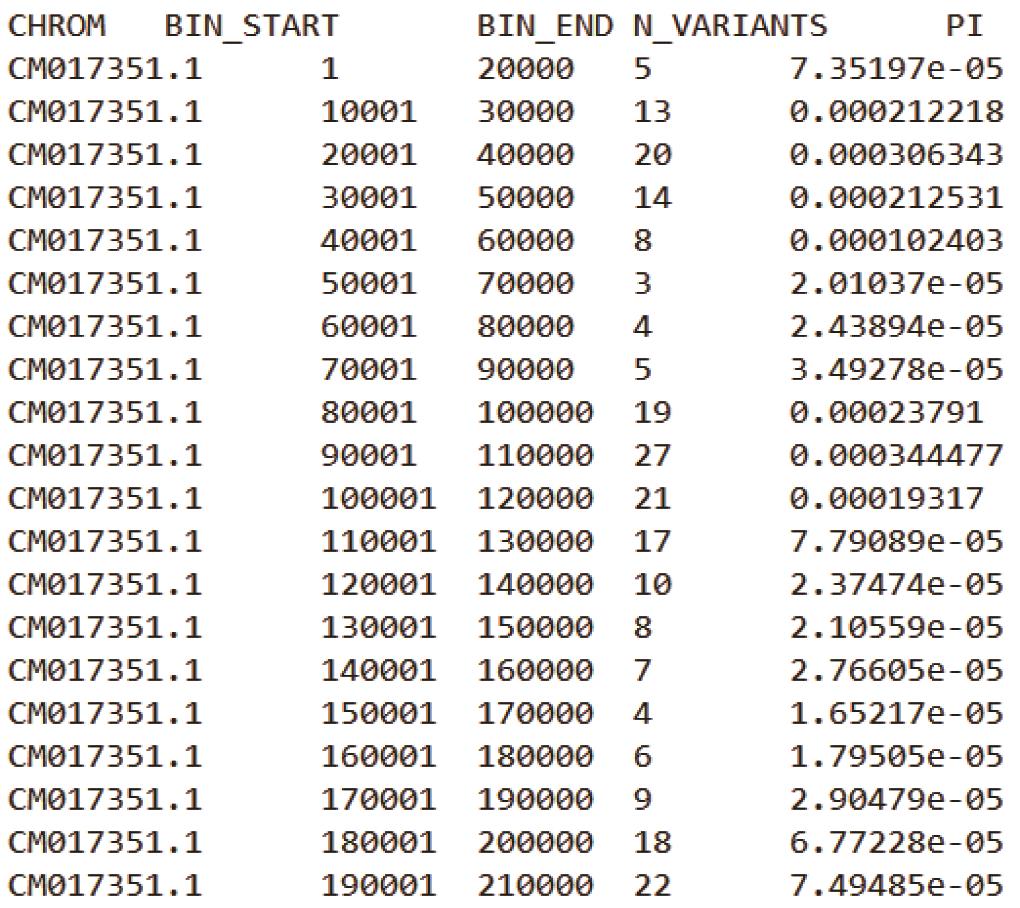
图3. 20K滑动窗口的θπ结果文件(10K 步长)
注:每个结果文件第一行为表头信息,CHROM为基因组中染色体/Scaffold编号;BIN_START 为每个窗口的起始坐标,BIN_END为终止坐标;N_VARIANTS表示该窗口内所包含的SNPs数目;PI为核苷酸多样度。第二个图中可以看到VCFtools以20K窗口大小和10K的步长在基因组上进行滑动和计算。
- 解析结果文件
以‘Samples.20K.windowed.pi’文件为例,使用R软件进一步对全基因组窗口化的核苷酸多样度计算结果进行解析。
Diversity<-read.table(file="Samples.20K.windowed.pi", header=T)
注:在R环境下将结果文件“Samples.20K.windowed.pi”读入‘Diversity’变量中用于后续统计分析;
mean(Diversity$PI)
注:计算该群体全基因组窗口化核苷酸多样度平均值
sd(Diversity$PI)
注:计算该群体全基因组窗口化核苷酸多样度标准差
median(Diversity$PI)
注:计算该群体全基因组窗口化核苷酸多样度中位值
distr<-density(Diversity$PI);
plot(distr, main="Distribution of Nucleotide diversity", xlab="Bins of Windowed θπ (20K)", ylab="Frequency", col="skyblue2");
注:统计该群体全基因组窗口化核苷酸多样度的分布密度(图4)
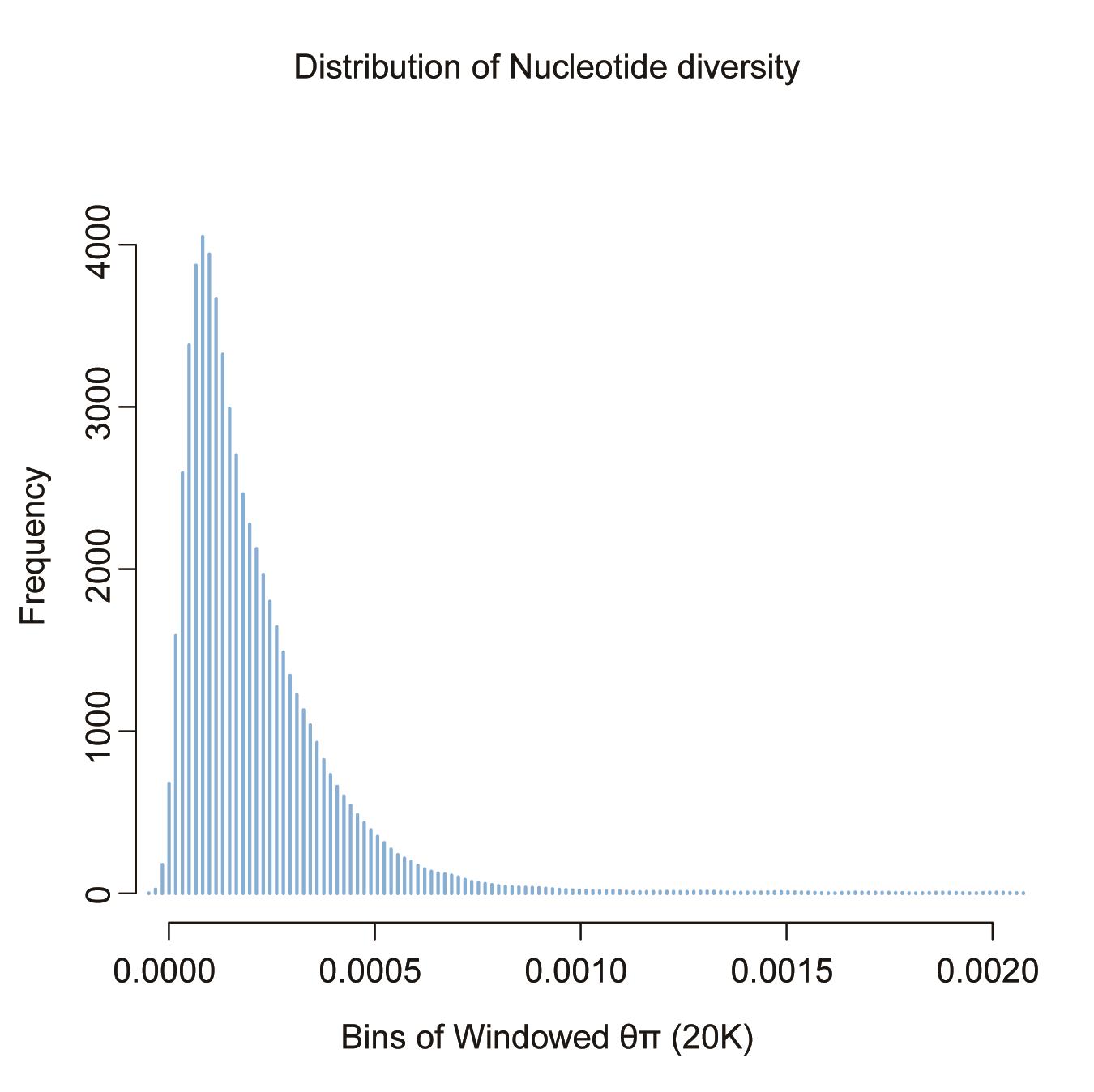
图4. 全基因组核苷酸多样度分布模式
除了从全基因组层面解析该群体的平均核苷酸多样度水平(平均值或者中位值)和分布模式外,还可以解析窗口化θπ在每条染色体/Scaffold上的分布和波动情况。θπ显著偏低的遗传区域暗示可能经历了自然选择作用。例如:从结果文件‘Samples.20K.windowed.pi’,中提取‘CM017351.1’染色体/Scaffold上的计算结果并解析核苷酸多样度在该遗传区域的分布和波动。
grep "^CM017351.1\s" Samples.20K.windowed.pi >> CM017351.1.20K.windowed.pi
注:从全基因组的结果文件中提取CM017351.1区域的结果用以分析
chr1<-read.table(file="CM017351.1.20K.windowed.pi", header=T)
注:读取该染色体区域的结果信息到变量‘chr1’中
plot(chr1$BIN_START,chr1$PI, ylab="Windowed θπ", xlab="Scans on CM017351.1", pch=20,type="h", col="gray");
abline(h=1.77778e-05,col="red2",lty=2);
注:图形化显示该染色体上窗口化θπ的分布和波动(图5)
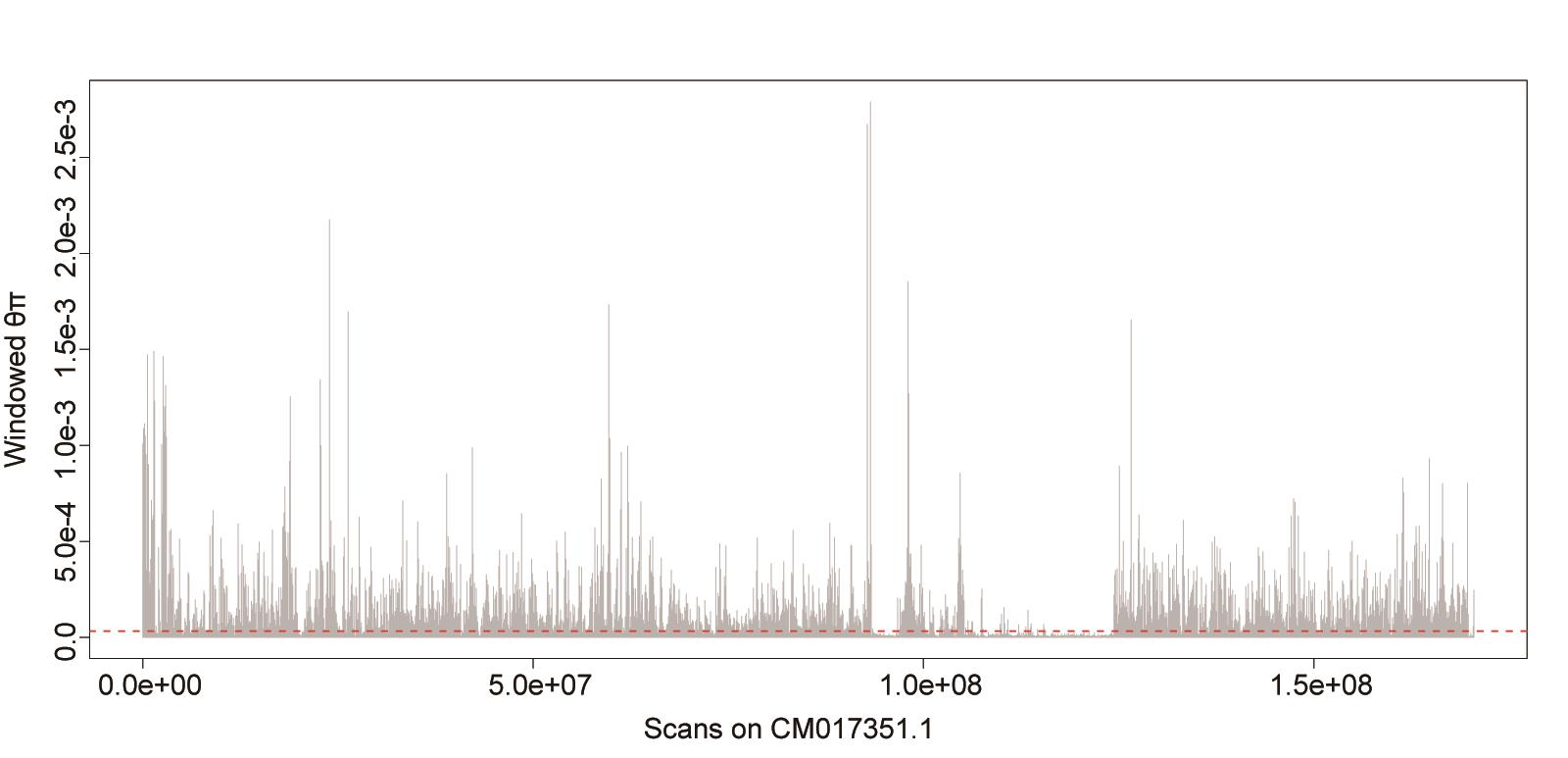
图5. CM017351.1号染色体上θπ的分布和波动
注:由图可见核苷酸多样度在染色体上并不是恒定不变,不同区域存在较大波动,反应了这些区域可能经历了不同的演化事件或显著差异的DNA遗传特性(如重组率和突变速率)。全基因组范围的核苷酸多样度符合正态分布,其平均值和标准差的大小往往反映了该种群基因组总体的遗传进化特征。然而,基因组中某些区域的多样度水平会急剧变小而偏离于基因组平均水平,统计上这些区域一般可以第一或第五分位数分别表征其对基因组偏离的显著性(1%和5%显著性差异)。它们往往反映了该区域可能经历了非随机的演化事件(例如自然选择);上图中,核苷酸多样度水平低于红色虚线(表征基因组第五分位数阈值)的遗传区域则可能经历了自然选择作用。
致谢
本工作得到了国家杰出青年科学基金项目(编号:31925006)和国家自然科学基金重大研究计划项目(编号:91731311)资助。
参考文献
- Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., Handsaker, R. E., Lunter, G., Marth, G. T., Sherry, S. T., McVean, G., Durbin, R. and Genomes Project Analysis, G. (2011). The variant call format and VCFtools. Bioinformatics 27(15): 2156-2158.
- Kuang, W.M., Hu, J.Y., Wu, H., Fen. X.T., Dai, Q.Y., Fu, Q.M., Xiao, W., Frantz, L., Roos, C., Nadler, T., Irwin, D.M., Zhou, L.C, Yang, X., Yu L. (2020) Genetic Diversity, Inbreeding Level, and Genetic Load in Endangered Snub-Nosed Monkeys (Rhinopithecus). Frontiers in Genetics. 11:1574-1583.
Copyright: © 2021 The Authors; exclusive licensee Bio-protocol LLC.