- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Identification and Monitoring of Nucleotide Repeat Expansions Using Southern Blotting in Drosophila Models of C9orf72 Motor Neuron Disease and Frontotemporal Dementia
Published: Vol 12, Iss 10, May 20, 2022 DOI: 10.21769/BioProtoc.4424 Views: 2364
Reviewed by: Oneil Girish BhalalaThomas MoensSébastien Gillotin
Original research article
The authors used this protocol in:

Sep 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Repeat expansion diseases, including fragile X syndrome, Huntington’s disease, and C9orf72-related motor neuron disease and frontotemporal dementia, are a group of disorders associated with polymorphic expansions of tandem repeat nucleotide sequences. These expansions are highly repetitive and often hundreds to thousands of repeats in length, making accurate identification and determination of repeat length via PCR or sequencing challenging. Here we describe a protocol for monitoring repeat length in Drosophila models carrying 1,000 repeat C9orf72-related dipeptide repeat transgenes using Southern blotting. This protocol has been used regularly to check the length of these lines for over 100 generations with robust and repeatable results and can be implemented for monitoring any repeat expansion in Drosophila.
Keywords: Southern blottingBackground
Repeat expansion diseases are a class of genetic disorders associated with the expansion of tandem repeat DNA sequences. The polymorphic nature and inherent instability of tandem repeats renders them prone to mutation, and as such they are one of the most abundant causes of variation in the human genome (Gymrek, 2017). The length and location of repeat sequences can vary, ranging from a single nucleotide to several nucleotides per repeat, situated within both coding and non-coding regions. There have been over 50 repeat expansion disorders identified, most of which primarily affect the central nervous system (Depienne and Mandel, 2021).
Repeat expansions in coding regions tend to be trinucleotide repeats, resulting in abnormally long repetitive amino acid sequences within proteins. In contrast, expansions in the UTRs or introns of genes are more varied in terms of sequence and how they confer toxicity. The repeat sequence itself can vary in length, the longest reported being a dodecamer in the 5’UTR of the CSTB (cystatin B) gene, which results in progressive myoclonic epilepsy type 1 (EPM1) (Lalioti et al., 1997). Moreover, whilst GC-rich repeat expansions in the 5’UTR tend to cause loss of function toxicity via epigenetic mechanisms such as persistent DNA hypermethylation, expansions situated within 3’UTRs and introns more frequently lead to a gain of function toxicity through RNA toxicity or polypeptide synthesis via repeat associated non-AUG (RAN) translation. An example of a repeat expansion that both reduces gene expression and elicits RNA and polypeptide repeat toxicity is the intronic GGGGCC expansion in the C9orf72 gene, the leading genetic cause of frontotemporal dementia (FTD) and motor neuron disease (MND) (Renton et al., 2011; DeJesus-Hernandez et al., 2011). Research using genetic models, including fruit flies, has determined that, whilst the C9orf72 expansion does reduce gene expression and cause haploinsufficiency, the main driver of toxicity in this case is the production of dipeptide repeat proteins (DPRs) from non-canonical RAN translation of the repeat itself. As translation occurs in all frames and from both sense and antisense RNA, five different DPRs are produced: glycine-alanine (GA), glycine-proline (GP), glycine-arginine (GR), alanine-proline (AP), and proline-arginine (PR) (Mori et al., 2013). These DPRs have been shown to aggregate in patient brains and spinal cord and are toxic in multiple model systems (May et al., 2014; Mizielinska et al., 2014; Zhang et al., 2014; Moens et al., 2019; West et al., 2020).
To further our understanding of expansion disorders and the mechanisms underpinning them, a range of transgenic model systems have been developed. For each expansion disorder, there is usually a consensus as to what constitutes a pathogenic repeat length. For example, the intronic hexanucleotide GGGGCC repeat in the C9orf72 gene is normally under 30 repeats in healthy individuals, whereas in those who develop disease it comprises over 500 and often thousands of repeats (Renton et al., 2011; DeJesus-Hernandez et al., 2011). In West et al. (2020), we developed the first Drosophila models expressing each DPR, individually and without repeat RNA, at over 1,000 repeats. To do this, alternative coding sequences were designed to produce the DPRs independently of the GGGGCC repeat (Callister et al., 2016). However, the transgene is still highly repetitive, GC-rich, and over 6000 bp in length, making accurate genotyping of repeat length via PCR or sequencing challenging. Furthermore, tandem repeats are increasingly unstable in a length dependent manner (Depienne and Mandel, 2021), and repetitive sequences introduced into bacteria and animal models are known to retract or be excised completely through generations (Bichara et al., 2006; Ryan et al., 2019). Therefore, it is important to be able to determine the length of repeat sequences in transgenic models and monitor them to check for retractions. The most common method, and widely considered gold standard, is Southern blotting.
Southern blotting is a highly sensitive technique used to detect specific DNA sequences in a blood or tissue sample. It involves the digestion of DNA with site-specific restriction endonucleases to isolate the genomic region of interest, followed by the separation of DNA fragments by size using gel electrophoresis and transfer onto a porous, positively charged nylon or nitrocellulose membrane via capillary action. The DNA of interest is detected using molecular hybridisation with specific nucleic acid probes, which are tagged with either radioisotopes or non-isotopic reagents (e.g., to facilitate chemiluminescent detection) (Southern, 2006). Whilst Southern blotting has been largely replaced by modern sequencing techniques and is no longer widely used, it remains an essential technique for researchers working with long and repetitive DNA sequences. Repeat sequences have always proved technically challenging for sequence alignment and assembly, and this is exacerbated when the repeat exceeds the sequencing fragment length (typically 350–500 bp). Moreover, traditional PCR amplification libraries have an inherent GC-bias, which can result in under-representation of reads from repeat expansions, a large proportion of which have 100% GC content (Treangen and Salzberg, 2012). These inaccuracies can lead to under-estimation of repeat sizes and mischaracterisation of repeat expansions (Rajan-Babu et al., 2021). It has been reported that PCR-based techniques used to detect the C9orf72 expansion in FTD/MND patients are unreliable, with both a high false-positive and false-negative rate (Akimoto et al., 2014). Therefore, whilst there are promising developments in long-read sequencing analysis, at present Southern blotting is considered the gold standard for genotyping GC-rich repeat expansions. Here we have developed and refined a protocol for the Southern blotting of repeat sequences in Drosophila. We optimized this protocol using Drosophila C9orf72 DPR models published in West et al. (2020), but it can be applied to other repeat expansions with alterations to the procedure required only for the oligonucleotide probes and restriction endonucleases.
Materials and Reagents
Whatman® 3MM filter paper (Whatman®, GE Healthcare, catalog number: WHA30306185)
Extra thick blotting paper (2.5 mm) (ThermoFisher Scientific, Thermo ScientificTM, catalog number 88605)
AmershamTM HybondTM-N 0.45 µm pore, neutral nylon membrane (GE Amersham, catalog number: RPN203N)
15 mL Falcon tubes (e.g., Corning® 15 mL centrifuge tubes, Sigma-Aldrich, Merck, catalog number: CLS430790)
50 mL Falcon tubes (e.g., Corning® 50 mL centrifuge tubes, Sigma-Aldrich, Merck, catalog number: CLS430829)
1.5 mL microfuge tubes (e.g., Eppendorf® Safe-Lock microcentrifuge tubes, Sigma-Aldrich, Merck, catalog number: T9661)
Plastic film (e.g., clingfilm) (Scientific Laboratory Supplies, catalog number: FIL1003)
Acetate sheets (Rapid Electronics Ltd, Diacel, catalog number: 34-0303)
Petri dish or weighing boat (for picking heads as shown in Figure 1, e.g., small square weighing boat, Sigma-Aldrich, Merck, catalog number: Z708542-500EA)
Phenol-chloroform-isoamylalcohol (25:24:1) saturated with 10 mM Tris, pH 8.0, 1 mM EDTA (Sigma-Aldrich, Merck, catalog number: P3803)
Chloroform (analytical reagent grade) (Sigma-Aldrich, Merck, catalog number: 366927)
Ethanol (for molecular biology) (Sigma-Aldrich, Merck, catalog number: 51976)
Ethidium Bromide, Molecular Biology-grade Aqueous Solution (500 mg/mL) (Sigma-Aldrich, Merck, catalog number: E1385)
Concentrated (36.5–38.0%) hydrochloric acid (HCl) (Sigma-Aldrich, Merck, catalog number: H1758)
Sodium chloride (NaCl) (analytical reagent grade) (Sigma-Aldrich, Merck, catalog number: S9888)
Sodium hydroxide (NaOH) pellets (anhydrous) (Sigma-Aldrich, Merck, catalog number: S5881)
Sodium citrate (Sodium citrate tribasic dihydrate) (Sigma-Aldrich, Merck, catalog number: C8532)
TRIS hydrochloride (TRIS HCl) pH 8.0 (Sigma-Aldrich, Merck, catalog number: 10812846001)
Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, Merck, catalog number: L3771)
Ethylenediaminetetraacetic acid (EDTA) (Merck, Sigma-Aldrich, catalog number: E9884)
Drosophila stocks to be tested (50–60 flies per genotype (see Notes for applying this to other tissues/protocols), including a negative wild-type control such as Canton S or Oregon R). Wild-type flies can be acquired from Bloomington Drosophila Stock Centre (BDSC, Indiana University) (Canton S: BDSC 9515, Oregon R BDSC 2376) or other stock centre. Transgenic stocks used in our example were generated by microinjection of pUAST-DPR-EGFP into VK00005 embryos by Cambridge Microinjection Facility [see West et al. (2020); available upon request].
Plasmids used to generate the transgenic fly lines, for use as positive controls [e.g., pUAST-DPR(1000)], see West et al., 2020, available upon request
Dry ice
Nuclease free water (molecular biology grade) (Sigma-Aldrich, Merck, catalog number: W4502)
Proteinase K (ThermoFisher Scientific, Thermo ScientificTM, catalog number: EO0491), divide into 1 mL aliquots and store at -20°C
Restriction enzymes to excise DNA of interest from the genome with appropriate buffer; for our example, we used Dde1 (New England Biolabs, catalog number: R0175) and NlaIII (New England Biolabs, catalog number: R0125), along with CutSmart® buffer (New England Biolabs, catalog number: B7204).
TBE Buffer (Tris-borate-EDTA) (10×) (ThermoFisher Scientific, Thermo ScientificTM, catalog number: B52)
Agarose for molecular biology (Sigma-Aldrich, Merck, catalog number: A9539)
6× loading dye containing bromophenol blue (New England Biolabs, catalog number: B7021S)
DNA Molecular Weight Marker II, DIG-labeled (Roche, Merck, catalog number: 11218590910)
1 kb Plus DNA Ladder (NEB, catalog number: N3200)
Probes specific to target sequence [The probes used in West et al., (2020), synthesised by Eurofins UK, are listed in the notes section of this protocol].
DIG easy hyb (Roche, Merck, catalog number: 11603558001)
Salmon sperm DNA (Agilent Technologies, catalog number: 201190), divide into aliquots of 300 µL and 150 µL and store at -20°C.
DIG Wash and Block Buffer Set (Roche, Merck, catalog number: 15857 62001), aliquot DIG block (e.g., 50 mL aliquots) and store at -20°C for long term (one aliquot can be stored for up to 3 months at 4°C).
Anti-Digoxigenin-AP, Fab fragments (Roche, Merck, catalog number: 11093274910, RRID:AB_2734716), store at 4°C
Ultrapure water (for example from Milli-Q® Purification System)
CDP-Star® chemiluminescent substrate (Roche, Merck, catalog number: CAS160081-62-9), store at 4°C
70% Ethanol (see Recipes)
Depurination solution (see Recipes)
Gel denaturing solution (see Recipes)
Gel neutralizing solution (see Recipes)
20× SSC stock (see Recipes)
10% SDS Solution (see Recipes)
2× SSC, 0.1% SDS (see Recipes)
0.5× SSC, 0.1% SDS (see Recipes)
0.1× SSC, 0.1% SDS (see Recipes)
Positive controls (see Recipes)
Genomic extraction buffer (see Recipes)
1× TBE (see Recipes)
Ladder mix (see Recipes)
1× DIG block (see Recipes)
1× maleic acid buffer (see Recipes)
1× maleic acid wash buffer (see Recipes)
1× detection buffer (see Recipes)
Equipment
Pipettes and tips (P1000, P200, P20, P10, and P2)
Fine paintbrush (e.g., RS PRO Thin 6.4 mm paintbrush, RS Components Ltd, catalog number: 2379190)
Vortex mixer (e.g., Vortex-Genie® 2 mixer, Sigma-Aldrich, Merck, catalog number: Z258415)
Fume hood
pH meter
Standard 700W–1000W microwave
Standard laboratory microfuge (e.g., Sigma 1-14 Microfuge, Sciquip, catalog number: 90616)
Tabletop mini microfuge (e.g., SciSpin MINI Microfuge, Sciquip, catalog number: SS-6050)
Wheel tube rotator (e.g., Cole-ParmerTM Stuart TM Rotator Disk, Fisher Scientific, catalog number: 11496548)
PowerPacTM Basic Power Supply (Bio-Rad, catalog number: 1645050)
Sub-Cell GT Electrophoresis Cell with 20-well combs and gel casting tray (15 × 15 cm) (Bio-Rad, catalog number: 1704402)
Clean plastic trays and sandwich boxes for incubating the membrane and gel, and assembling the Southern blot (Figure 2)
Large sandwich container (e.g., 250 × 150 mm) to use as the basin for Southern blotting apparatus (Figure 2)
Chemiluminescence and fluorescence imaging system (e.g., G:box imaging unit, Syngene)
HB pencil
Scissors/guillotine for cutting filter paper and membrane
UV Transilluminator (e.g., dual wavelength (302/365) 8W transilluminator LM-20, VWR, catalog number: 732-4388)
Standard boiling water bath (e.g., VWR, SBB Aqua 5 plus, catalog number: 462-0171) or hot block capable of reaching >100°C
Standard hybridisation oven (e.g., HB-1000 Hybridiser, VWR, catalog number: 732-4300)
Glass hybridisation bottles to fit in hybridisation oven (e.g., 35 × 150 mm hybridisation bottles, VWR, catalog number: 732-4350)
Flat edged forceps (e.g., S MurrayTM Stainless Steel Forceps L325/01, Fisher Scientific, catalog number: 12342158)
Rocker (e.g., Cole-ParmerTM StuartTM See-Saw Rocker, catalog number: 10470655)
Software
Imaging software for imaging the blot, for example: GeneSys and GeneTools (Syngene, https://www.syngene.com/support/software-downloads/)
Procedure
Prepare Reagents
Before starting the protocol ensure that you have prepared the positive controls, ladder mixture, and all buffers and solutions described in the recipes section. All recipes, excluding 1× DIG block and 1× detection buffer, which must be made up from 10× on the day of use, can be made in advance and stored for at least 1 year. Proteinase K must be added fresh to the genomic extraction buffer immediately before use.
DNA extraction from Drosophila heads (whole flies or other tissues could be used, but we find that using heads gives a better signal due to the lack of contaminants from, for example, gut contents).
DAY 1
Collect 50–60 flies (yielding ~25–30 µg genomic DNA), per genotype, in 15 mL Falcon tubes and place on dry ice. In addition to test samples, collect wild-type flies, for example Canton S or Oregon R, to use as a negative control.
To separate the fly heads, place 3–4 pieces of dry ice into a 50 mL Falcon tube and place the 15 mL Falcon tube containing the flies inside the 50 mL Falcon tube (see Figure 1A). Do not put the lid back on the 50 mL Falcon tube. Vortex at top speed until heads detach (~30 s) (see Video 1). Keep on dry ice at all times when not vortexing.
 Video 1. Detaching
Video 1. DetachingDrosophila heads by vortexing.Pick heads and transfer them to a 1.5 mL microcentrifuge tube on dry ice. Heads can be picked manually using whichever method is easiest. Our recommended method is to tip the decapitated flies onto a petri dish lined with filter paper sitting in a large tray or weighing boat containing dry ice (see Figure 1B). Use a fine paintbrush to pick up the heads (see Figure 1C) and transfer them to a 1.5 mL tube on dry ice. This procedure is easier when everything is kept as cold as possible.
Defrost proteinase K and add to genomic extraction buffer (see Recipes).
Homogenize the heads by “squishing” with a 1–200 µL pipette tip filled with genomic extraction buffer (1 µL per head) without expelling the liquid (sufficient buffer will be expelled during the process to efficiently homogenize the heads without them floating around) (~10 s). Then expel the remaining extraction buffer and put in the hybridisation oven (or another incubator) and set to 57°C. Incubate overnight.
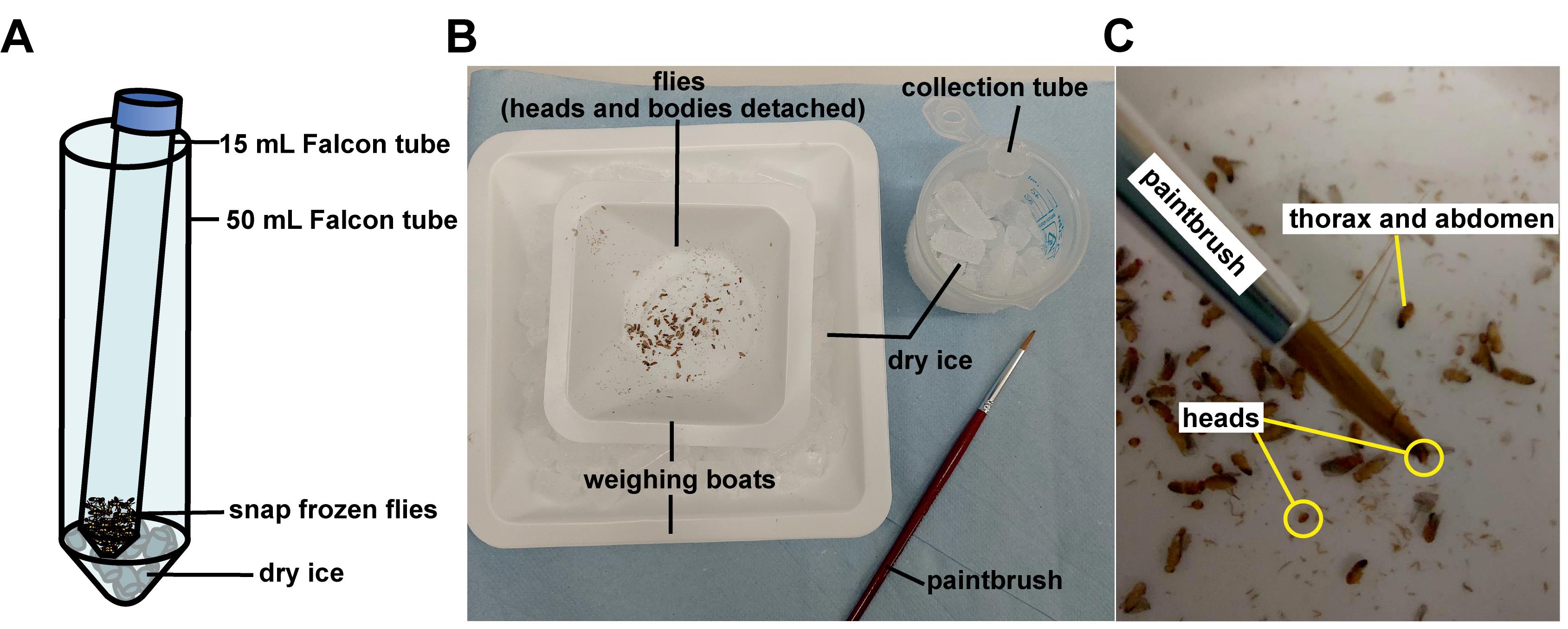
Figure 1. Example apparatus for collecting Drosophila heads. A. Apparatus for detaching Drosophila heads using a vortex mixer (see also Video 1). B. Apparatus for picking heads using a paintbrush. C. Heads detached from the rest of the fly.
DAY 2
The following day, in a fume hood, add one volume (equivalent to the volume used in step 5) phenol-chloroform-isoamylalcohol (25:24:1) and incubate at room temperature for 15 min with rotation (using a wheel tube rotator, for example).
Centrifuge for 5 min at ~13,000 × g.
Transfer the aqueous (top) layer to a new tube.
Add one volume of 100% chloroform to the recovered aqueous phase from step 8, incubate at room temperature for 15 min with rotation (using a wheel tube rotator, for example), and repeat steps 7–8.
Recover DNA with standard ethanol precipitation:
Add 1 mL of 100% ethanol, invert for one minute, and place at -20°C overnight.
The following day, spin at 13,000 × g for 10 min and discard supernatant.
Wash the pellet with 800 µL of 70% ethanol.
Centrifuge at 13,000 × g for 5 min and discard supernatant.
Leave the pellet to air dry for 15–30 min at room temperature and then resuspend in 100 µL of nuclease free water.
Allow to dissolve overnight at 4°C or 1 h at room temperature. At this point DNA can be frozen and stored at -20°C until ready to move on to section C.
DNA digests
DAY 4
Note: Restriction enzymes were chosen to give efficient digestion of genomic DNA without cutting the target sequence. For our example, DdeI and NlaII were chosen as they efficiently cut the vector used to insert our construct into the fly genome, without cutting the DPR sequence [see Figure 2 and West et al., (2020)]. Whilst the approximate cutting frequency of each enzyme within the genome should be considered (for example, NlaIII cuts a 4 bp site so has a cutting frequency of ~44), the most critical factor is that the enzyme does not cut your target sequence. The enzymes used in our example are listed in Materials and Reagents and detailed in Table 1. Digests are carried out in a large volume of 500 µL to allow complete digestion of genomic DNA. All the DNA from a 50–60 head extraction is used (~25–30 µg DNA).
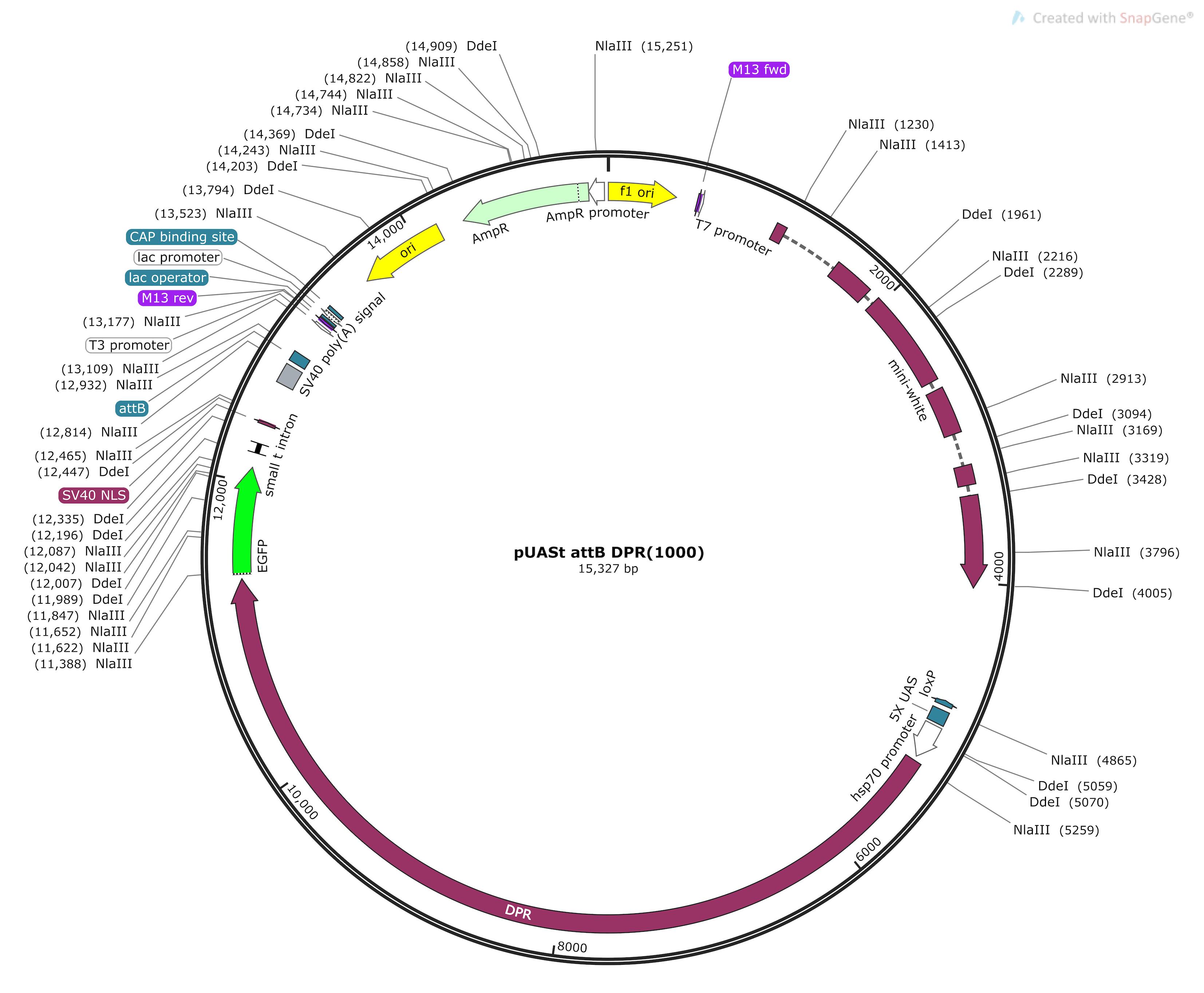
Figure 2. Plasmid map indicating cut sites of restriction enzymes NIaIII and Dde1. pUASt-DPR1000-GFP plasmid map showing mini-white element, DPR sequence, and EGFP tag. Restriction sites for NlalII and Dde1 are indicated (made using Snapgene Viewer).Set hybridisation oven (or other incubator) to required temperature for your restriction enzymes.
Prepare reaction mixtures (example shown in Table 1).
Mix by pipetting and settle contents by centrifugation in a tabletop microfuge (10 s at 2,680 × g).
Incubate in optimal conditions for your chosen enzymes (in our example, overnight at 37°C).
DAY 5
Recover DNA with standard ethanol precipitation:
Add 1 mL of 100% ethanol, invert for one minute, and place at -20°C overnight.
DAY 6
The following day, spin at 13,000 × g for 10 min and discard supernatant.
Wash the pellet with 800 µL of 70% ethanol.
Centrifuge at 13,000 × g for 5 min and discard supernatant.
Leave the pellet to air dry for 15-30 min at room temperature.
Resuspend the pellet in 17 µL of nuclease free water, mix well, and centrifuge briefly in a tabletop microfuge (10 s at 2,680 × g) to ensure all the DNA is at the bottom of the tube.
Allow the DNA to dissolve for 2–3 days at 4°C before running the Southern blot.
Table 1. DNA digestion mixture example
Volume (µL) DNA 100 Cutsmart buffer (10×) 50 NIaIII 2 Dde1 2 Nuclease free water 346 Total Volume 500
Blotting samples
DAY 7 (2–3 days after Day 6)
Make an agarose gel:
Mix 130 mL 1× TBE with an appropriate mass of agarose to make a gel of the percentage required to resolve your construct (see Table 2 for a guide to gel percentages and resolving capability based on product size; in our example, we use 1% for a 6 kb product).
Boil the TBE agarose mix in a microwave.
Add 0.5 µg/mL ethidium bromide and swirl to mix.
Pour into a 15 × 15 cm casting tray using a 20 well comb and leave to set.
Prepare samples for electrophoresis:
Defrost the ladder mix and 10 ng/µL positive control stock.
Add 0.5 µL of positive control to 9.5 µL of nuclease free water and 2 µL of 6× loading dye to give a total volume of 12 µL.
For test samples and negative controls prepared in sections B and C, add 3 µL of 6× loading dye to give a total volume of 20 µL, which is the maximum that can be loaded into the wells.
Prepare to run the gel:
Place the gel into the tank, fill the tank with 1× TBE, and remove the comb.
Load 7 µL of ladder mix, 12 µL of positive control, and 20 µL of samples to individual wells. It can be beneficial to leave blank wells either side of the positive control to avoid false positives resulting from spill over between wells.
Electrophorese DNA samples at 100 V for long enough to give good separation between ladder bands around target DNA (for a 6 kb product this is approximately 2.5 h).
Table 2. Guide for % agarose in TBE gel based on expected product size
Agarose % (w/v) Resolution 0.50% 1,000–30,000 bp 0.70% 800–12,000 bp 1.00% 500–10,000 bp 1.20% 400–7,000 bp 1.50% 200–3,000 bp 2.00% 50–2,000 bp Image the gel using a gel documentation system such as a G-box. This is useful to ascertain whether the DNA has digested fully (digests should appear as smears down the lanes) and to refer to later if the signal is weak in certain lanes when imaging the final blot. It is also helpful to use the UV transilluminator to help trim the blot.
The gel can be trimmed smaller, using the ladder as a guide to ensure DNA of interest is not removed, if desired. Cut one corner of the gel to help identifying the gel orientation and transfer the gel into a clean plastic container.
Depurinate the gel: add enough depurination solution to cover the gel and shake slowly at room temperature on a see-saw rocker or orbital shaker, until the bromophenol blue in the loading dye turns yellow or for no longer than 10 min.
Pour off the depurination solution and briefly rinse the gel in distilled water before the next step.
Denature the DNA in the gel: add enough gel denaturing solution to cover the gel and incubate at room temperature for 30 min with shaking.
Neutralize the gel: pour off denaturing solution and replace with the same volume of neutralising solution. Incubate at room temperature with shaking for 30 min.
Pour off the neutralising solution and replace with the same volume of 20× SSC. Incubate for 20 min at room temperature with shaking before blotting. This helps to equilibrate the gel and remove background.
Meanwhile, prepare 3 sheets of Whatman 3 MM paper, a sheet of Hybond-N 0.45 µm pore nylon membrane, and 15 sheets of extra thick blotting paper, all cut to the same size as the gel. Also cut 3 strips of 3MM paper slightly wider than the gel, and longer than the length of the support to use as wicks (see Figure 2).
At this point, you can cut a corner off the membrane to allow the blot to be orientated after hybridisation, and put a pencil mark on the side that will face the gel to identify the DNA side.
Clean flat edged forceps using 70% ethanol to remove alkaline phosphatase before using them to handle the nylon membrane (alkaline phosphatase reacts with chemiluminescent substrates used in the detection process).
Assemble the transfer as shown in Figure 3. For this step, 20× SSC can be reused.
Place a small plastic container (the bridge/support) upside down inside a larger container to form the basis of your transfer apparatus.
Place the longer strips of filter paper over the bridge/support to act wicks, wicking the 20× SSC in the buffer reservoir up into the gel. Pour a little 20× SSC in to wet the wicks.
Place the gel onto the wick covered bridge/support (we typically place our gels well side up; however, this is not important as transfer can occur in either orientation), followed by the nylon membrane (pencil mark down facing the gel and with the cut corner of the membrane matching the cut corner of the gel, to aid with orientation). Be careful to remove all bubbles (gently with gloved fingers) between the gel and membrane.
Follow with the sheets of 3MM paper and extra thick blotting paper. To avoid short circuiting the transfer, make sure the buffer cannot bypass the gel and membrane as it is drawn up to the thick blotting paper, by ensuring the wicks do not touch the blotting paper above the gel and membrane.
Place a weight (~100–300 g, for example a small plastic container filled with enough water to cover the base but not spill) on top of the paper and leave the gel overnight at room temperature to transfer. Ensure the wicks are immersed and the weight is level. If the room temperature is warm and you have problems with evaporation of the 20× SSC, cover the apparatus with plastic film to prevent this.
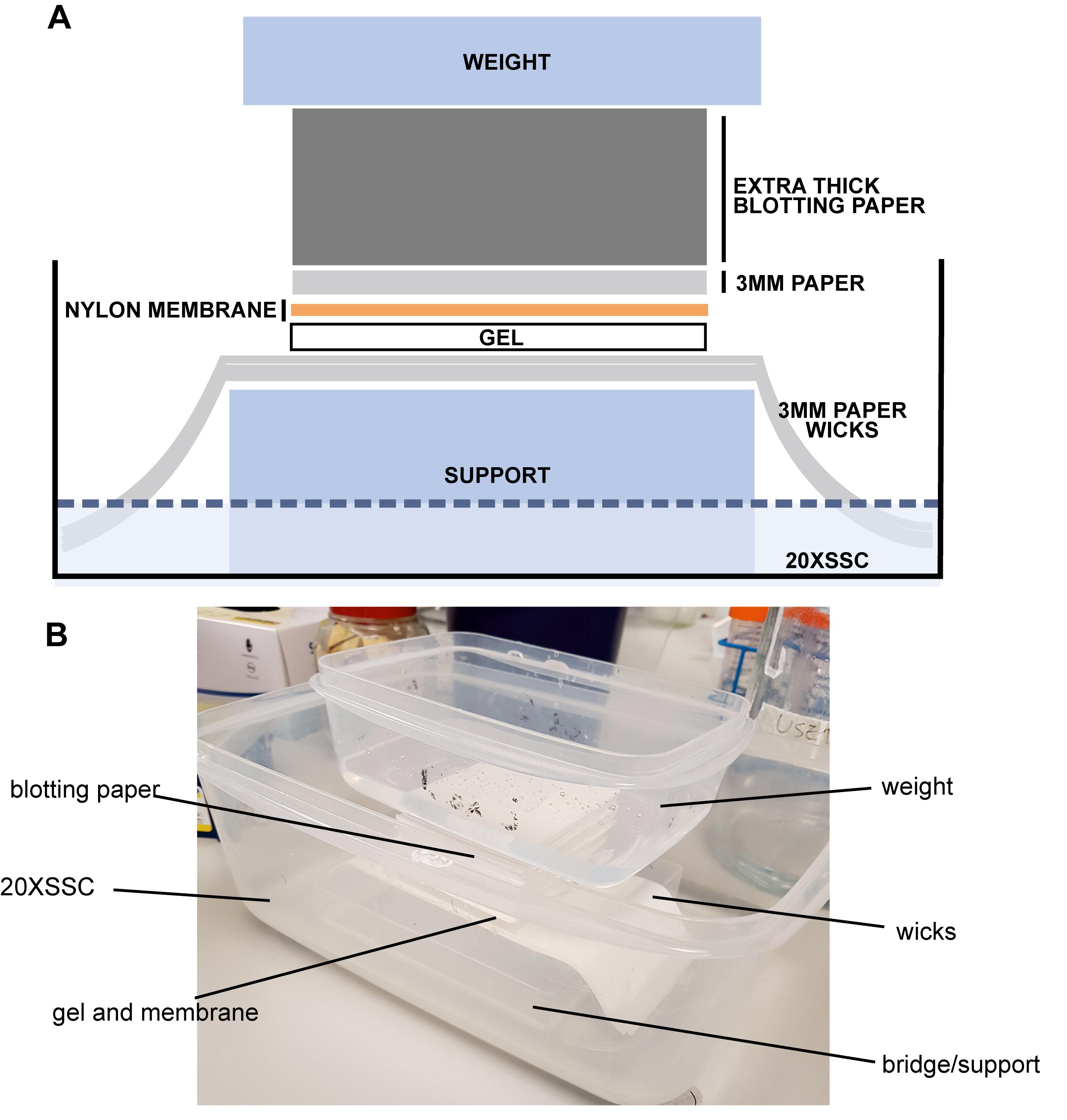
Figure 3. Southern blotting assembly. A. Schematic. B. Photo. Place a small plastic container upside down in a larger plastic container to form a bridge. Place the longer strips of filter paper over the container to allow the 20× SSC in the buffer reservoir to wick up into the gel. Pour a little 20× SSC in to wet the wicks. Place gel on the bridge, followed by nylon membrane, being careful to remove all bubbles between the gel and membrane. Carefully place the sheets of 3MM paper and extra thick blotting paper on top. Place a weight (for example, a plastic container filled with water) on top of the paper and leave the gel overnight to transfer. Ensure that the wicks are covered and the weight level. If you are worried about evaporation you can cover in plastic film.Hybridisation
DAY 8
The following day, set the boiling water bath/hot block to 100°C and the hybridisation oven to 42°C.
Place 30 mL DIG easy hyb at 42°C to prewarm. Defrost 300 µL salmon sperm DNA.
Disassemble the blot (keep 20× SSC for reuse) and gently wash the membrane in ~20 mL of 2× SSC.
Immobilise the DNA on the membrane using UV (302 nm, “Hi” intensity setting): place plastic film over the surface of the UV transilluminator, place the membrane DNA side down, turn on the illuminator, and leave to fix for 180 s (Figure 4). If using a UV crosslinker such as a Stratalinker, use a standard autocrosslinking setting at 1,200 µJ.
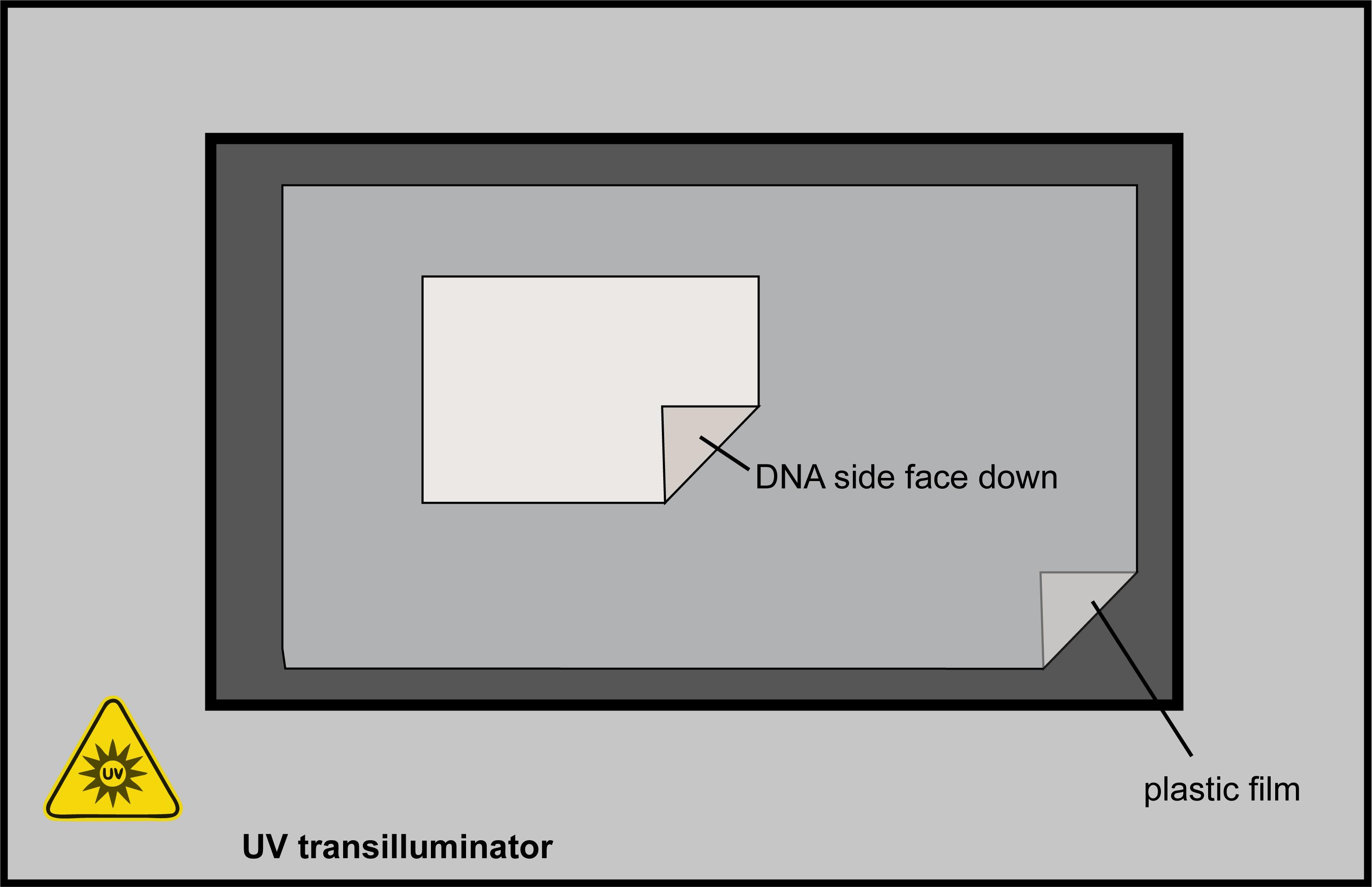
Figure 4. UV Fixing the membrane - schematic. Ensure you are wearing correct personal protective equipment for using UV. Place plastic film over the surface of the UV transilluminator and the membrane DNA side down on top of the plastic film. Turn on the UV and leave to fix for 180 s.Transfer to a hybridisation bottle (DNA side facing inwards; ensure the DNA side of the membrane is fully exposed and not folded over on itself) and add ~50 mL of 2× SSC to stop the membrane drying out*.
Note: * The membrane can be left in 2× SSC overnight, but if it is to be left longer before hybridisation, rinse the membrane twice for 10 min in ultrapure water, then gently air dry the membrane and store between sheets of 3MM paper for later hybridisation. This washing is crucial as it removes any traces of salt that may dry on the membrane and result in background signal.
Prepare pre-hybridisation solution: boil 300 µL of salmon sperm DNA stock to denature (this is 3,000 µg DNA) for 10 min and then place on ice to prevent re-annealing; add to 30 mL of prewarmed DIG easy hyb.
Pour off 2× SSC if using/place dry membrane in hybridisation bottle DNA side inwards and add the pre-hybridisation solution. Pre-hybridise with rotation for 4 h at 42°C.
Prepare hybridisation solution: defrost 150 µL (1,500 µg) salmon sperm DNA, prewarm 15 mL DIG easy hyb to 42°C; denature salmon sperm DNA as in step 6 and add this and 7.5 µL 10 ng/μL oligo probe stock to DIG easy hyb, giving a final salmon sperm DNA concentration of 100 µg/µL and probe concentration of 0.005 ng/µL.
Pour off pre-hybridisation solution and replace with hybridisation solution. Hybridise overnight at 42°C with rotation.
Detection
DAY 9
The following day, remove the hybridisation bottles from the oven and set to 65°C.
Put 2 × 50 mL aliquots of 2× SSC; 0.1% SDS and 1 × 50 mL 0.5× SSC; 0.1% SDS (and 50 mL 0.1× SSC; 0.1% SDS if using for optional extra final wash (step 7)) at 65 °C to prewarm. Defrost 10× DIG block and prepare 1× block (see Recipes).
Pour off the probe, and rinse the membrane in the hybridisation bottle with approximately 50 mL 2× SSC; 0.1% SDS.
Add 50 mL of prewarmed 2× SSC; 0.1% SDS to the membrane in the bottle and wash for 15 min at 65°C with rotation.
Replace with 50 mL of fresh prewarmed 2× SSC; 0.1% SDS and wash for 15 min.
Replace with 50 mL of prewarmed 0.5× SSC; 0.1% SDS and wash for 15 min.
Optional extra wash (if background is high and your signal is weak): replace with 50 mL of prewarmed 0.1× SSC; 0.1% SDS and wash for 15 min.
Pour off the last of the hybridisation wash solutions and rinse the bottle out with ~50 mL 1× maleic acid buffer (shake vigorously before use).
Transfer the membrane to a clean dish and incubate the membrane at room temperature with shaking for 2 min in maleic acid buffer.
Prepare the antibody solution by centrifuging the anti-digoxigenin-AP antibody for 15 min at 13,000 × g, to remove antibody complexes. Pipette antibody from the top to make 20 mL 1:20,000 anti-DIG in 1× DIG block.
Pour off the maleic acid buffer and incubate the membrane for 30 min at room temperature in 50 mL 1× DIG block with shaking.
Pour off the blocking solution and incubate the membrane at room temperature for 30 min in 20 mL antibody solution with shaking.
Wash the membrane twice (2 × 15 min, at room temperature, and with shaking) in approximately 50 mL 1× maleic acid wash buffer.
Prepare 1× detection buffer (see Recipes). Equilibrate the membrane for 5 min in 20 mL 1× detection buffer. Leave in detection buffer at room temperature until ready to image.
Place the membrane DNA side up on a sheet of clean acetate. Add 1–2 mL (to cover the membrane) CDP-Star® chemiluminescent substrate, distributed evenly over membrane. Place another sheet of clean acetate over the top, sandwiching the membrane in between. Be careful to eliminate bubbles as you place the second sheet of acetate over the membrane. Proceed to imaging immediately.
Image the membrane using the G-Box or equivalent system initially for 1 min to check the blot has worked (should see the ladder) and then again at the highest quality.
For enhanced detection of weak bands, highlight the area of interest and set the auto exposure time based on these bands. Image again. This may take up to 30 min using the G-Box system listed here. See Figure 5 for an example final Southern blot image.
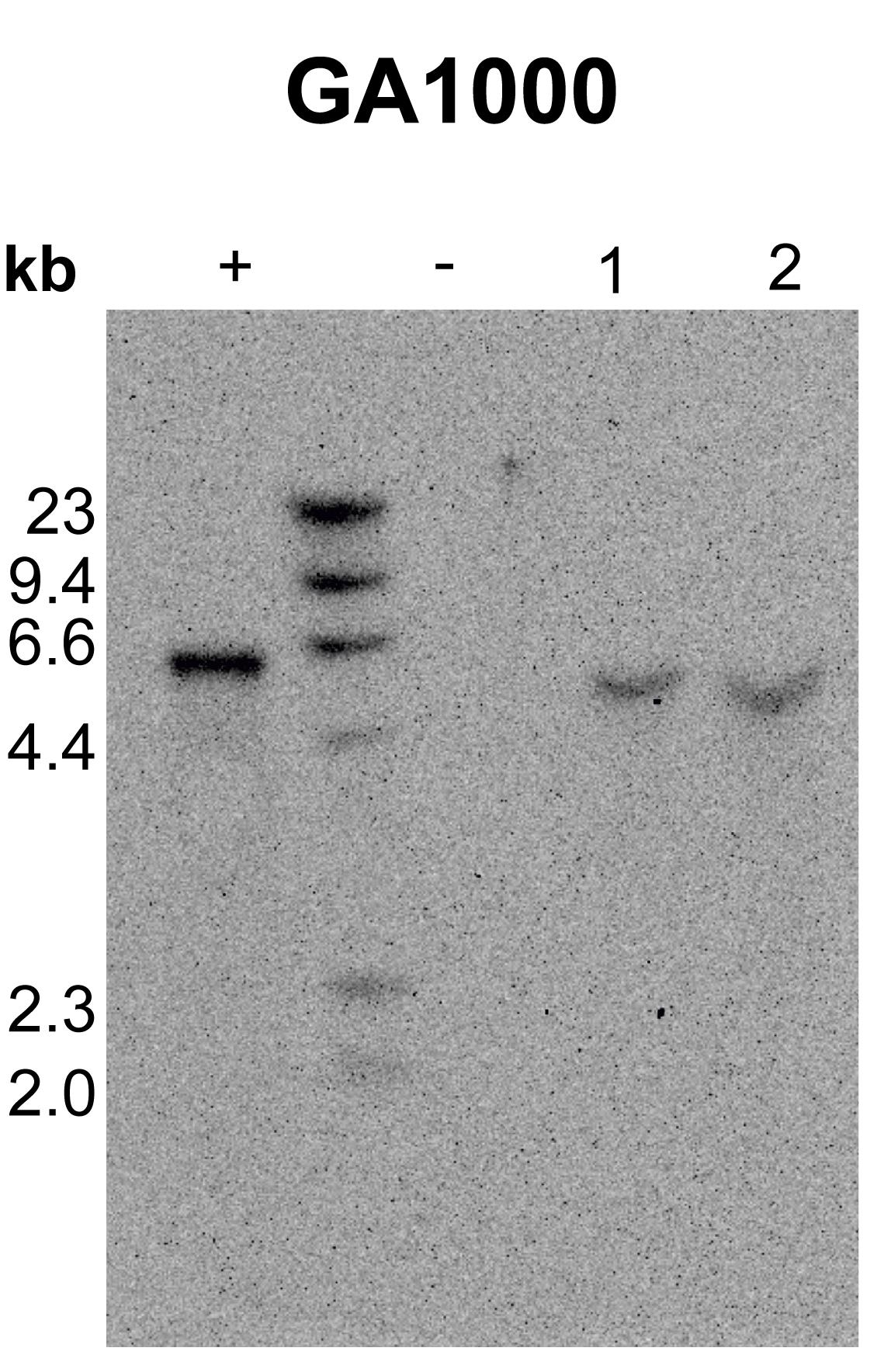
Figure 5. An example Southern blot used to length check UAS-GA1000-EGFP fly lines. A 6 kb band corresponds to 1000 dipeptide repeats. Lanes in order: positive control (+, DNA from wild-type flies spiked with 1000 repeat linearized plasmid DNA), ladder, negative control (-, DNA from wild-type flies), and two independent GA1000 lines (1 and 2). This blot was imaged for 30 min.
Notes
We found that extraction from 50-60 heads was optimal (25–30 µg genomic DNA), but if using other genomic DNA extraction protocols, or are extracting DNA from other tissues, this may require optimizing. We recommend a minimum of 5 µg genomic DNA should be used.
Different DIG ladders can be used if the DNA of interest is of a shorter/longer repeat length.
If the target DNA is different to the examples given, optimisation of the following will be required:
Gel percentage and running time
Amounts of positive control loaded, based on the intensity relative to samples
For further optimisation of background reduction, we recommend adding an additional stringent wash after hybridisation, as noted in procedures section F step 7. A third 15 min wash in maleic acid wash buffer after antibody incubation (section F step 12) may also help reduce high background levels.
For Southern blotting of Drosophila generated in West et al. (2020) the following probes were used:
GA probe: DIG-GGCAGGAGCTGGAGCTGGCGCAGGAGCTGGTGCTGGG-DIG
GR probe: DIG-AGGCAGAGGTCGTGGGAGAGGCAGGGGTCGCGGACGTGGA-DIG
AP probe: DIG-AGCACCAGCACCGGCGCCAGCTCCAGCACCAGCACCC-DIG
PR probe: DIG-AGACCCCGTCCTCGTCCTCGTCCAAGACCAAGGCCGAGGC-DIG
The probe design will vary depending on your target sequence. Generally, oligonucleotide probes should be between 18 and 50 bases to balance yield and specificity. An optimal GC content is between 40–50%, and computational analysis is recommended to optimize specificity.
Recipes
Unless otherwise stated, buffers and solutions should be kept at room temperature for up to one year.
Genomic extraction buffer
25 mM NaCl, 10 mM Tris-HCl pH 8.2, 1 mM EDTA, and Proteinase K 200 μg/mL
Store buffer without Proteinase K at room temperature; proteinase K should be stored at –20°C in 1 mL aliquots and added to the appropriate volume of buffer immediately before use.
Positive controls
Wild-type Drosophila DNA spiked with 157 ng of linearized plasmid containing target repeat sequence per 1 µg genomic DNA, to produce the equivalent of haploid DNA. Keep a stock of 10 ng/µL at -20°C.
Ladder mix
1:1 DIG-labelled DNA marker II: 1 kb plus DNA ladder, plus 1× loading dye (the visible ladder is used as a reference for the size of the DNA fragments when the gel is trimmed after electrophoresis). Keep a stock of this mixture at -20°C.
70% Ethanol (make up in nuclease free water)
Make up in nuclease free water
1× TBE
Make up in ultrapure water
Depurination solution
0.25 M HCl
Gel denaturing solution
0.6 M NaCl, 0.2 N NaOH
17.53 g of NaCl, 4 g NaOH in 500 mL water
Gel neutralizing solution
1.5 M NaCl, 0.5 M Tris-HCl pH 8.0
43.8 g NaCl, 30.30 g Tris, in 500 mL water plus HCl to pH (approx. 30 mL)
20× SSC stock
3 M NaCl, 300 mM Sodium citrate pH 7.4
175.3 g NaCl, 88.2 g of sodium citrate per L distilled water
10% SDS Solution
50 g sodium dodecyl sulfate in a total volume of 500 mL water
2× SSC, 0.1% SDS
50 mL of 20× SSC, 5 mL of 10% SDS in a total volume of 500 mL of distilled water
0.5× SSC, 0.1% SDS
12.5 mL 20× SSC, 5 mL of 10% SDS in a total volume of 500 mL of distilled water
0.1× SSC, 0.1% SDS
2.5 mL 20× SSC, 5 mL of 10% SDS in a total volume of 500 mL of distilled water
This can be used for a final 15 min stringent wash if your blot has a lot of background, and the signal is weak.
1× DIG block (prepared on the day)
Dilute 10× DIG block (DIG Wash and Block Buffer Set) in 1× maleic acid buffer
1× maleic acid buffer
Dilute 10× malic acid buffer (DIG Wash and Block Buffer Set) in distilled water
1× maleic acid wash buffer
Dilute 10× malic acid wash buffer (DIG Wash and Block Buffer Set) in distilled water
1× detection buffer (prepared on the day)
Dilute 10× detection buffer (DIG Wash and Block Buffer Set) in distilled water
Acknowledgments
We thank Dr Sara Rollinson for her help and expertise with optimising this protocol.
This work was supported by an Alzheimer’s Society fellowship awarded to RJHW (AS-JF-16b-004 (510)), The Royal Society (RGS\R2\212216), a Medical Research Council studentship awarded to JLS and a Jean Corsan Foundation studentship awarded to NSH. This protocol is derived from our previous work (West et al., 2020; doi: 10.1186/s40478-020-01028-y).
Competing interests
The authors declare no competing interests
References
- Akimoto, C., Volk, A. E., van Blitterswijk, M., Van den Broeck, M., Leblond, C. S., Lumbroso, S., Camu, W., Neitzel, B., Onodera, O., van Rheenen, W., et al. (2014). A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet 51(6): 419-424.
- Bichara, M., Wagner, J. and Lambert, I. B. (2006). Mechanisms of tandem repeat instability in bacteria. Mutat Res 598(1-2): 144-163.
- Callister, J. B., Ryan, S., Sim, J., Rollinson, S. and Pickering-Brown, S. M. (2016). Modelling C9orf72 dipeptide repeat proteins of a physiologically relevant size. Hum Mol Genet 25(23): 5069-5082.
- DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., Nicholson, A. M., Finch, N. A., Flynn, H., Adamson, J., et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72(2): 245-256.
- Depienne, C. and Mandel, J. L. (2021). 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am J Hum Genet 108(5): 764-785.
- Gymrek, M. (2017). A genomic view of short tandem repeats. Curr Opini Genet Dev 44: 9-16.
- Lalioti, M. D., Scott, H. S., Buresi, C., Rossier, C., Bottani, A., Morris, M. A., Malafosse, A. and Antonarakis, S. E. (1997). Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 386(6627): 847-851.
- May, S., Hornburg, D., Schludi, M. H., Arzberger, T., Rentzsch, K., Schwenk, B. M., Grasser, F. A., Mori, K., Kremmer, E., Banzhaf-Strathmann, J., et al. (2014). C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128(4): 485-503.
- Mizielinska, S., Gronke, S., Niccoli, T., Ridler, C. E., Clayton, E. L., Devoy, A., Moens, T., Norona, F. E., Woollacott, I. O. C., Pietrzyk, J., et al. (2014). C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345(6201): 1192-1194.
- Moens, T. G., Niccoli, T., Wilson, K. M., Atilano, M. L., Birsa, N., Gittings, L. M., Holbling, B. V., Dyson, M. C., Thoeng, A., Neeves, J., et al. (2019). C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol 137(3): 487-500.
- Mori, K., Arzberger, T., Grasser, F. A., Gijselinck, I., May, S., Rentzsch, K., Weng, S. M., Schludi, M. H., van der Zee, J., Cruts, M., et al. (2013). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126(6): 881-893.
- Rajan-Babu, I. S., Pen, J. R. J., Chiu, R., Li, C. K., Mohajeri, A., Dolzhenko, E., Eberle, M. A., Birol, I., Friedman, J. M., Study, I., et al. (2021). Genome-wide sequencing as a first-tier screening test for short tandem repeat expansions. Genome Medicine 13(1).
- Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., Schymick, J. C., Laaksovirta, H., van Swieten, J. C., Myllykangas, L., et al. (2011). A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 72(2): 257-268.
- Ryan, S., Hobbs, E., Rollinson, S. and Pickering-Brown, S. M. (2019). CRISPR/Cas9 does not facilitate stable expression of long C9orf72 dipeptides in mice. Neurobiol Aging 84: 8.
- Southern, E. (2006). Southern blotting. Nature Protocols 1(2): 518-525.
- Treangen, T. J. and Salzberg, S. L. (2012). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13(1): 36-46.
- West, R. J. H., Sharpe, J. L., Voelzmann, A., Munro, A. L., Hahn, I., Baines, R. A. and Pickering-Brown, S. (2020). Co-expression of C9orf72 related dipeptide-repeats over 1000 repeat units reveals age- and combination-specific phenotypic profiles in Drosophila. Acta Neuropathol Commun 8(1): 19.
- Zhang, Y. J., Jansen-West, K., Xu, Y. F., Gendron, T., Bieniek, K., Lin, W. L., Sasaguri, H., Caulfield, T., Hubbard, J., Daughrity, L., et al. (2014). Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128(4): 505-524.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sharpe, J. L., Harper, N. S. and West, R. J. H. (2022). Identification and Monitoring of Nucleotide Repeat Expansions Using Southern Blotting in Drosophila Models of C9orf72 Motor Neuron Disease and Frontotemporal Dementia. Bio-protocol 12(10): e4424. DOI: 10.21769/BioProtoc.4424.
Category
Neuroscience > Nervous system disorders > Neurodegeneration
Neuroscience > Nervous system disorders > Animal model
Biochemistry > DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.