- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification of the Bacterial Amyloid “Curli” from Salmonella enterica Serovar Typhimurium and Detection of Curli from Infected Host Tissues
Published: Vol 12, Iss 10, May 20, 2022 DOI: 10.21769/BioProtoc.4419 Views: 3233
Reviewed by: Andrea PuharAlberto J. Martin-RodriguezAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Microbiologists are learning to appreciate the importance of “functional amyloids” that are produced by numerous bacterial species and have impacts beyond the microbial world. These structures are used by bacteria to link together, presumably to increase survival, protect against harsh conditions, and perhaps to influence cell-cell communication. Bacterial functional amyloids are also beginning to be appreciated in the context of host-pathogen interactions, where there is evidence that they can trigger the innate immune system and are recognized as non-self-molecular patterns. The characteristic three-dimensional fold of amyloids renders them similar across the bacterial kingdom and into the eukaryotic world, where amyloid proteins can be undesirable and have pathological consequences. The bacterial protein curli, produced by pathogenic Salmonella enterica and Escherichia coli strains, was one of the first functional amyloids discovered. Curli have since been well characterized in terms of function, and we are just starting to scratch the surface about their potential impact on eukaryotic hosts. In this manuscript, we present step-by-step protocols with pictures showing how to purify these bacterial surface structures. We have described the purification process from S. enterica, acknowledging that the same method can be applied to E. coli. In addition, we describe methods for detection of curli within animal tissues (i.e., GI tract) and discuss purifying curli intermediates in a S. enterica msbB mutant strain as they are more cytotoxic than mature curli fibrils. Some of these methods were first described elsewhere, but we wanted to assemble them together in more detail to make it easier for researchers who want to purify curli for use in biological experiments. Our aim is to provide methods that are useful for specialists and non-specialists as bacterial amyloids become of increasing importance.
Keywords: CurliBackground
The bacterial cell surface appendages termed curli were first discovered in an E. coli strain that was isolated from horse manure. The authors named the surface structures “curli” because of their curved appearance (Olsén et al., 1989). At nearly the same time, similar surface structures were observed in Salmonella enterica serovar Enteritidis, where they were termed thin aggregative fimbriae (Collinson et al., 1991). The curli and thin aggregative fimbriae had many unique properties, including a requirement for treatment with 90% formic acid (FA) to break the fibers apart into monomeric proteins that could be resolved by SDS-PAGE gel. A landmark study published in 1998 revealed that curli and thin aggregative fimbriae, including the genes required for biosynthesis, were virtually interchangeable between S. enterica and E. coli (Romling et al., 1998). With the publication of the genome sequence of S. Typhimurium LT2, the “curli” nomenclature was chosen for both species. In terms of function, curli are key proteins in the formation of S. enterica and E. coli biofilms, with a primary role in “short-range” cell-cell interactions leading to aggregation (Romling et al., 1998). This aggregation and biofilm formation has been linked to improved persistence and survival in the face of environmental insults (Anriany et al., 2001; Solano et al., 2002; Scher et al., 2005; Ryu and Beuchat, 2005; White et al., 2006). It is quite unique for a surface structure like curli to be conserved between these related, but long diverged species; we have proposed that curli and biofilm formation plays a key role in survival of these enteric bacteria in the environment as they cycle between their hosts. Their conservation throughout S. enterica and E. coli speaks to their importance in the lifecycle of both bacterial species.
Curli are now known to be ‘functional amyloids’. Another landmark paper showed that CsgA, the curli monomer, has self-polymerization properties that are hallmark of amyloids (Chapman et al., 2002). This explained some of the unique, biochemically resistant physical properties of curli that were observed upon their initial purification. The characteristic 3-D structure of amyloids has been termed cross-beta with regular, repeated β-sheets or strands that run perpendicular to the fiber axis. The speculation with curli is that when CsgA monomers are stacked upon each other to form fibers, there is a central non-polar core that would run along the length of curli fibers, leading to extreme stability (Collinson et al., 1999). Assembly of fibers occurs outside of the cell, where unfolded CsgA monomers pass through the outer membrane through the CsgG pore and then snap into their more rigid cross-beta structure (Evans and Chapman, 2014). Although amyloids are notoriously difficult to work with and are not easily amenable to protein crystallography because of the non-uniform fiber-fiber interactions, some structural features have been determined through the use of CsgA-specific peptides (Szulc et al., 2021). Perhaps the most striking analysis performed to date revealed that CsgA “curli” have structural similarity to the amyloid-beta fibrils that are characteristic of Alzheimer’s disease, with both sharing a characteristic structural fold called the steric β-zipper (Perov et al., 2019).
The role of curli in host-pathogen interactions has recently been clarified. Several early studies described curli in the context of host expression (Bian et al., 2000; Olsén et al., 1998; Sjobring et al., 1994), but for a long period, we thought them to be purely an “environmental” factor. Pioneering work by Çagla Tükel, Andreas Baumler, and colleagues identified purified curli fibers as potent stimulators of the innate immune system, where they interact with Toll-like receptor 2 (TLR-2) (Tükel et al., 2005), TLR1/2 and TLR1/2/9 due to the presence of extracellular DNA in complex with curli (Tükel et al., 2010; Tursi and Tükel, 2018). Systemic presentation of curli had a profound impact on autoimmunity in host species, leading to increased presence of circulating anti-dsDNA antibodies, stimulation of nod-like receptors (i.e., NLRP3), and the inflammasome (Gallo et al., 2015; Rapsinski et al., 2015). Most recently, we showed that curli are expressed by S. enterica after oral infection of mice, inside the large intestine, in both acute and long-term infection models (Miller et al., 2020). In these experiments, production of curli by S. enterica cells inside the host led to increased levels of autoimmunity and early signatures of arthritis in the knee joints of infected mice. The impact of curli was tied to the ability of S. enterica to invade host cell epithelium and cause inflammation, indicating that curli must access the lymphoid tissues surrounding the intestine to have negative effects. Many aspects of curli expression in vivo still need to be clarified.
In this manuscript, we present a series of step-by-step protocols with pictures showing how to purify these important bacterial surface structures.
Materials and Reagents
Disposable Culture Tubes 16 × 100 mm (Fisherbrand, catalog number: 14-961-29)
DifcoTM Luria-Bertani broth (BD Biosciences, catalog number: 244620)
Polystyrene disposable Petri dishes 150 mm × 15 mm (VMR International, catalog number: 25384-326).
Cotton tipped applicator, 6 inch (Puritan, catalog number: 1495992B)
Precleaned Microscope Slides (Fisherbrand, catalog number: 12-550-343)
2 mL Safe Lock Tubes (Eppendorf, catalog number: 054027)
5 mm stainless steel bead (Qiagen, catalog number: 69989)
30 mL Oak Ridge round-bottom tubes (Thermo Scientific Nalgene, catalog number: 3115-0030)
Disposable 5ml syringe (BD Biosciences, catalog number: 309647)
Precision Glide Needle (BD Biosciences, catalog number: B305196)
0.22 µm Nitrocellulose membrane (FroggaBio, catalog number: TM300)
Filter paper (Bio-Rad, catalog number: 1703965)
1 ply Kimwipes 11 × 21 cm (KimTech Science Brand, Kimberly-Clark, Code: 34155)
BactoTM Tryptone (BD Biosciences, catalog number: 211705)
DifcoTM Agar, Bacteriological (BD Biosciences, catalog number: 214530)
Tris-HCl pH 8 (Invitrogen, catalog number: 15-568-025)
Dimethyl sulfoxide (DMSO, Sigma-Aldrich, catalog number: D-1435, CAS number: 67-68-5)
Trizma base (Sigma-Aldrich, CAS number: 77-86-1)
Hydrochloric Acid (Bio Basic Canada Inc. CAS number: 7647-01-0)
RNase A, Protease-Free, Highly Purified, Bovine Pancreas (Sigma-Aldrich, CAS number: 9001-99-4)
Deoxyribonuclease I from Bovine Pancreas (Sigma-Aldrich, CAS number: 9003-98-9)
MgCl2·6H2O (Bio Basic Canada Inc. CAS number: 7791-18-6)
Lysozyme from Chicken Egg White (Sigma-Aldrich, CAS number: 12650-88-3)
Sodium dodecyl sulfate (Bio Basic Canada Inc. catalog number: 151-21-3)
40% Acrylamide/Bis Solution 29:1 (Fisher Bioreagents, catalog number: BP1408-1)
Ammonium persulfate (Sigma-Aldrich, CAS number: 7727-54-0)
TEMED (Thermo Scientific, catalog number: 17919)
Sterile distilled water
β- mercaptoethanol (Sigma-Aldrich, LOT number: SHBJ8714; catalog number M6250)
Ethyl Alcohol 95% Vol (Commercial Alcohols by Greenfield Global, Item number: P016EA95)
Glycine (Bio Basic Canada Inc. catalog number: 56-40-6)
BLUelf Prestained Protein Ladder (FroggaBio, catalog number: PM008-0500)
Glycerol (Bio Basic, GB0232, CAS number: 56-81-5)
Bromophenol Blue (Sigma-Aldrich, CAS number: 34725-61-6)
NaCl (Bio Basic, DB0483, CAS number: 7647-14-5)
Tween 20 (Sigma-Aldrich, CAS number: 9005-64-5)
Methanol (Fisher Bioreagents, catalog no: BP1105-4)
IRDye 680RD goat anti-rabbit IgG (Mandel Scientific, ERP No: LIC-926-68071)
Nitro-BT (NBT) (Fisher BioReagentsTM, CAS number: 298-83-9)
5-Bromo-4-chloro-3-indolyl phosphate (BCIP) (Fisher BioReagentsTM, CAS number: 298-83-9)
Formic Acid 98-100% (Millipore Sigma, CAS number: 64-18-6)
1,1,1,3,3,3-Hexafluoro-2-propanol; Hexafluoroisopropanol (HFIP) (Sigma-Aldrich, CAS number: 920-6611)
InvitrogenTM NovexTM WedgeWellTM 4 to 20%, Tris-Glycine, 1.0 mm, Mini Protein Gel, 10-well (Thermo Fisher Scientific, catalog number: XP04200PK2)
InvitrogenTM iBlotTM 2 Transfer Stacks, nitrocellulose, mini (Thermo Fisher Scientific, catalog number: IB23002)
Infected tissue samples
N-cetyl-N,N,N,-trimethyl ammonium bromide (CTAB) (Sigma-Aldrich, CAS number: 57-09-0)
Phenol/chloroform/isoamyl alcohol (pH 7.7–8.3) (49.5:49.5:1) (Sigma-Aldrich, catalog number 77618)
Chloroform (Sigma-Aldrich, CAS number: 67-66-3)
MSB broth (see Recipes)
YESCA broth (see Recipes)
T agar (see Recipes)
2× SDS-PAGE sample buffer (100 mL) (see Recipes)
10× SDS-PAGE running buffer (1,000 mL) (see Recipes)
10× Towbin buffer or Transfer buffer (1,000 mL) (see Recipes)
10× TBS (1,000 mL) (see Recipes)
1× TBST (see Recipes)
12% separating gel (2 gels) (see Recipes)
5% stacking gel (2 gels) (see Recipes)
AP buffer (Alkaline Phosphatase) (500 mL) (see Recipes)
NBT/BCIP developing solution (see Recipes)
Equipment
Water bath
Autoclave vacuum steam sterilizer (GETINGE, model: 533LS)
Biological Safety Cabinet (Therma Electron Corporation, model: 1284)
Water Jacketed CO2 Incubator (Therma Electron Corporation, model: 3110)
Sonicator (BRANSON, DIGITAL SONIFIER 450)
Tissue Homogenizer (Retsch, high-speed mixer mill MM400)
Superspeed Centrifuge (Thermo Scientific, SORVALL EVOLUTION RC)
SDS-PAGE gel big apparatus (Bio-Rad, PROTEAN II xi Cell, model: 1651814)
InvitrogenTM Mini Gel Tank (Thermo Fisher Scientific, model: A25977)
Lyophilizer (FTS SystemTM, Duro-DryTM MP, model: FD2085C0000)
Freezer (Panasonic, SANYO, model: MDF-U76VC)
Weighing balance (METTLER TOLEDO, model: B2002-S)
Trans-Blot SD semi-dry transfer cell (Bio-Rad Laboratories)
InvitrogenTM iBlotTM 2 Gel Transfer Device (Thermo Fisher Scientific, model: IB21001)
Odyssey CLx imaging system and Image Studio 4.0 software package (Li-Cor Biosciences, model: 9140)
DeNovix DS-11 FX Microvolume spectrophotometer/fluorometer
Procedure
A mutant strain used for curli purification was S. enterica serovar Typhimurium 14028-3b ∆bcsA. This is a strain of wild-type S. Typhimurium 14028 that we engineered to produce more curli by introducing a csgD promoter mutation from S. enterica serovar Enteritidis 27655-3b (White and Surette, 2006). csgD encodes the master biofilm regulator in S. enterica and E. coli (Gerstel et al., 2003; Brombacher et al., 2006). The other modification was to introduce a single-gene knockout of bcsA (White et al., 2006), which encodes cellulose synthase, responsible for making cellulose polymers at the cell surface of S. enterica and E. coli biofilm cells (Zogaj et al., 2001; Solano et al., 2002). The presence of cellulose causes other substances to become trapped in the extracellular matrix during the purification process. Furthermore, because curli and cellulose are difficult to separate (White et al., 2003), these carbohydrates can represent a significant proportion of the final purified product. The use of a cellulose-negative ∆bcsA strain avoids purification of these unnecessary contaminants.
The same procedure can be used to purify curli from strains of S. enterica or E. coli that produce cellulose; it is just important to note that cellulose will represent a proportion of the final weight of the “mature curli”. The tight association between curli and cellulose (White et al., 2003) will make it impossible to separate them.
Mature Curli Extraction and Purification
Bacterial Strain and culture conditionGrow strain S. Typhimurium 14028-3b ∆bcsA in 5 mL of Luria broth (1% salt) overnight at 37°C with shaking at 200 rpm.
Dip sterile swabs in this overnight culture and spread evenly onto 120 large (150 mm × 15 mm) T agar plates to ensure that a homogenous lawn of bacteria covers the entirety of each plate.
Incubate the inoculated plates at 28°C for 48 h.
Collect the bacterial cells and extracellular material by scraping the agar surfaces using microscope slides (Video 1) or a sterile cell scraper.
 Video 1. Scraping Plates
Video 1. Scraping PlatesSuspend the cell materials from each set of 10 plates in 20 mL of 10 mM Tris-HCl (pH 8) supplemented with RNase A and DNase I (to 0.1 mg/mL final concentration).
Vortex the suspended cell material until there are no visible clumps.
Transfer 1 mL of cell slurry into 2 mL Safe-Lock tubes containing a 5 mm stainless steel bead (Qiagen) (approximately 20 tubes per 10 Petri plates).
Place tubes in the mixer mill and homogenize at 30 Hz for 5 min in room temperature to break up the extracellular matrix.
Transfer and combine the homogenized cell materials into 30 mL Oak Ridge centrifuge tubes.
Lyse bacterial cells by sonication using the 3 mm probe with 10 × 30 s pulses at 30% amplitude with 2 min cooling on ice between pulses (Figure 1).

Figure 1. Reference image of sonication apparatus used for bacterial cell lysis. Cells are suspended in a 30 mL Oak Ridge centrifuge tube, which is embedded in ice within a 500 mL beaker. The sonicator probe is placed within the centrifuge tube approximately 5 mm beneath the surface of the liquid cell slurry.After sonication, add 2 M MgCl2 to a final concentration of 1 mM and incubate the mixture at 37°C for 20 min without shaking.
After incubation, add lysozyme to a final concentration of 1 mg/mL and incubate the mixture at 37°C for 40 min without shaking.
Add SDS to a final concentration of 1%, along with 0.1 mg/mL DNase I, and incubate this mixture overnight at 37°C without shaking.
The next day, centrifuge the mixture in 30 mL Oak Ridge centrifuge tubes at 25,000 × g and 4°C for 25 min.
For each tube, remove supernatant and resuspend the pellet in 10 mL of 10 mM Tris-HCl (pH 8), boil for 10 min, and centrifuge the mixture at 25,000 × g for 25 min. Repeat this step twice.
Resuspend the pellet with 10 mL of 10 mM Tris-HCl (pH 8) and add RNase A (0.1 mg/mL final conc.), DNase I (0.1 mg/mL final conc.) and Lysozyme (1 mg/mL final conc.) and incubate the mixture overnight at 37°C.
Note: This step can be repeated several times, if the mixture is highly viscous (i.e., looks like a clump of mucus), without any reduction in amount or quality of the final, purified material.
After the mixture has achieved a lower viscosity (i.e., becomes watery), centrifuge the mixture at 12,100 × g for 15 min.
Wash the pellet twice with 10 mL of 10 mM Tris-HCl (pH 8) by centrifugation at 12,100 × g for 15 min. Reduced viscosity is necessary for this step to work efficiently and can slightly vary from batch to batch.
Resuspend the pellet in 3 ml of 2× SDS-PAGE sample buffer and boil for 15 min (Crude protein sample).
Mature Curli Purification in SDS-PAGE Gel.
Prepare a 4 mm thick SDS-PAGE gel with 5% stacking gel and 12% separating gel (Figure 2A).
Load 3 mL of Crude protein sample from step 16 (see above) into the well of preparative SDS-PAGE gel (Figure 2B) and run continuously at 100V until all the dye in the sample runs through the bottom (Figure 2C). Pack around the bottom of the electrophoresis apparatus with ice to prevent the gel from over-heating (Figure 2B).

Figure 2. SDS-PAGE gel apparatus for curli purification from bacterial cells. (A) Reference image of gel casting system. (B) Three milliliters of crude protein sample loaded into the well, prior to SDS-PAGE. Note: pack the SDS-PAGE gel apparatus with ice or run the experiment in a 4°C cold room to prevent gel overheating. (C) Electrophoresis is performed until the sample loading dye reaches the bottom.Collect the insoluble material retained on the top of the well (Figure 3; arrow) using a 5 mL syringe with 18-gauge needle. Purified curli fibers will not enter the SDS-PAGE gel unless they are depolymerized with 90% FA (Collinson et al. 1991) or 100% HFIP (Zhou et al., 2013).

Figure 3. Collection of curli aggregates from the top of the gel after electrophoresis. The red arrow denotes the purified curli fibrils on the top of the well before collection. Using a syringe and an 18-gauge needle, the researcher can collect the white material from the top of the gel.Wash recovered insoluble material with 10 mL of sterile distilled water and sediment by centrifugation at 16,000 × g and 4°C for 10 min. Repeat this wash step three times.
Dissolve the pellet with 5 ml of 95% ethanol and centrifuge at 16,000 × g and 4°C for 10 min. Repeat this step twice.
Resuspend the pellet with 10 mL of sterile distilled water, transfer to suitable container (e.g., 30 mL Oakridge centrifuge tubes; Figure 4A), and freeze the mixture at -80°C.
Wrap the frozen tubes with 1 ply kimwipes, secure them with rubber band, and lyophilize for 24 h. White, flocculent material should be present at the bottom of each tube (Figure 4B).
Note: Tubes can be either wrapped with 1 ply kimwipes or with aluminum foil. If the tubes are wrapped with aluminum foil, make few holes for the proper lyophilization of frozen material. These holes allow the change of frozen material from solid to vapor state.
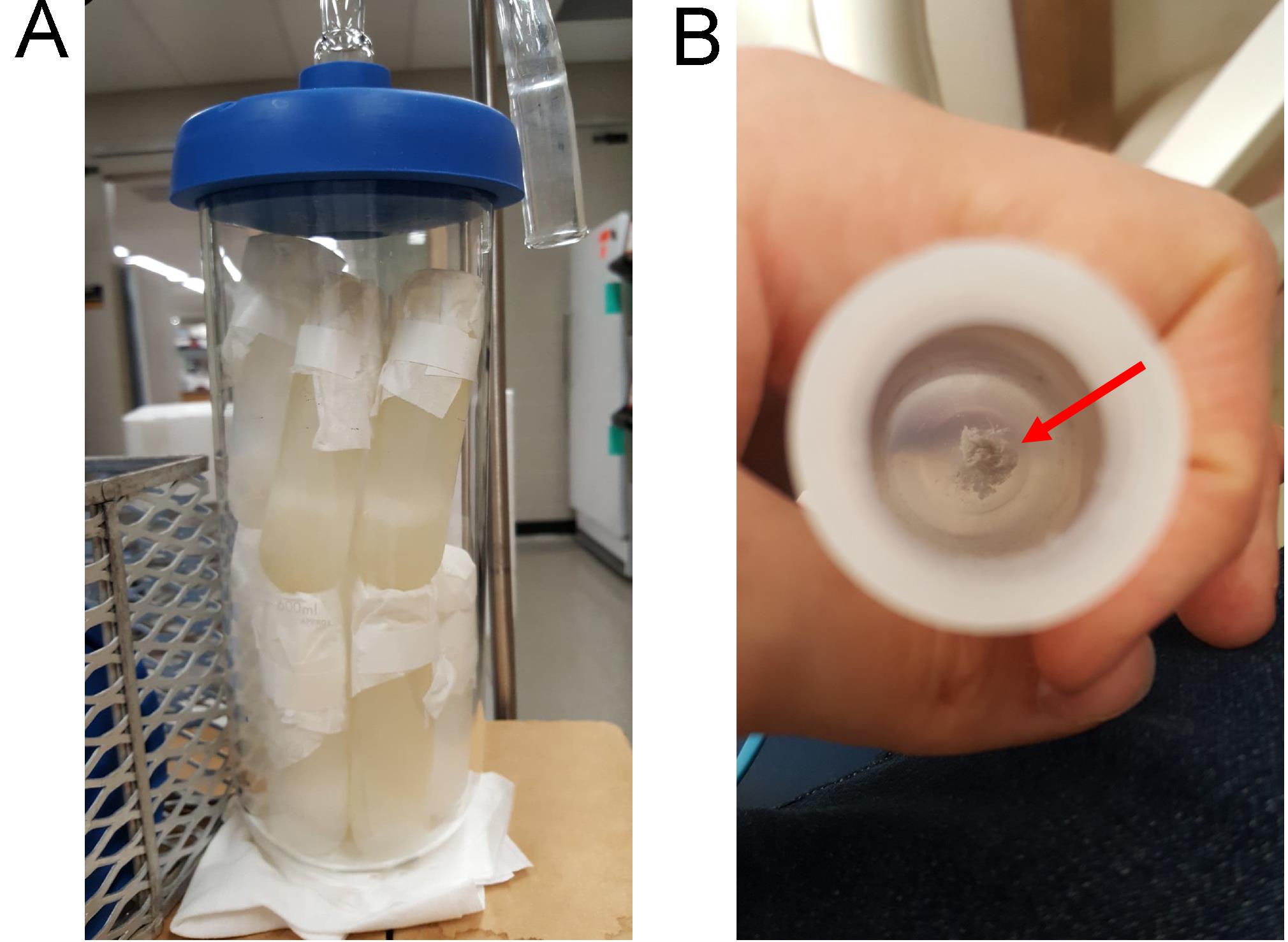
Figure 4. Lyophilization of purified curli aggregates. (A) The solution of purified curli in water was frozen at -80°C in 30 mL Oakridge centrifuge tubes, wrapped with lint free tissue, and lyophilized for 24 h. (B) White, flocculent material (red arrow) present in the bottom of each tube after lyophilization represents curli fibrils.Dissolve the lyophilized material in 2 mL of 0.2 M glycine (pH 1.5) and boil this mixture for 10 min to solubilize any Type I fimbriae that might be present (Müller et al., 1991).
Centrifuge the sample at 27,500 × g for 25 min.
Wash the pellets five times with sterile distilled water with centrifugation at 25,000 × g for 25 min.
Resuspend the pellet with 5 mL of distilled water and transfer the mixture into a pre-weighed glass vial.
Freeze the mixture at -80°C and lyophilize for 24 h.
After lyophilization, weigh the glass vial containing sample to obtain the rough sample weight. The final purified material (Figure 5) should be white to off-white in color and “more powdery” than the material described in step 6.

Figure 5. Quantitation and long-term storage of purified curli. All material collected after the final lyophilization step was transferred into pre-weighed, sterile glass vials, and the rough weight was calculated. This purified material can be stored at -20°C.Store the lyophilized material (Curli standard) at -20°C. Stock solutions are best prepared by weighing out a desired amount of purified curli into a glass vial, and resuspending in distilled water to achieve the desired concentration, such as 1 mg/mL. Once a stock solution is prepared, aliquot the mixture into Eppendorf tubes. Because purified curli fibers are insoluble in water, make sure to evenly mix the stock solution and use a wide-bore pipette to transfer the aliquot.
To prepare depolymerized curli, add 90% FA or 100% HFIP to the mature curli (purified curli from step 13).
Note: HFIP and FA are irritating and corrosive chemicals. Always use them under a chemical fume hood and wear appropriate protective equipment while handling. Store them in a well-ventilated, cool place.
For HFIP treatment, weigh 1 mg of curli, dissolve it in 1 mL of HFIP, and aliquot them into 0.5 mL Eppendorf tubes. For FA treatment, prepare 1 mg/mL of curli in sterile water, aliquot 10 µL of stock into 0.5 mL Eppendorf tubes, and add 92 µL of FA.
Immediately freeze the mixtures for 1 h at -80°C, make a hole in the lids (i.e., using a sterile needle), and lyophilize for 24 h.
Note: Samples containing acid may damage some lyophilizers.
After lyophilization, dissolve mature (from step 13) and FA/HFIP-treated or depolymerized curli (from step 15–16) with 30 µl of 1× SDS-PAGE sample buffer and load them into each SDS-PAGE gel lane.
Check the purity of mature and depolymerized curli by immunoblot analysis (Figure 6A and 6C).
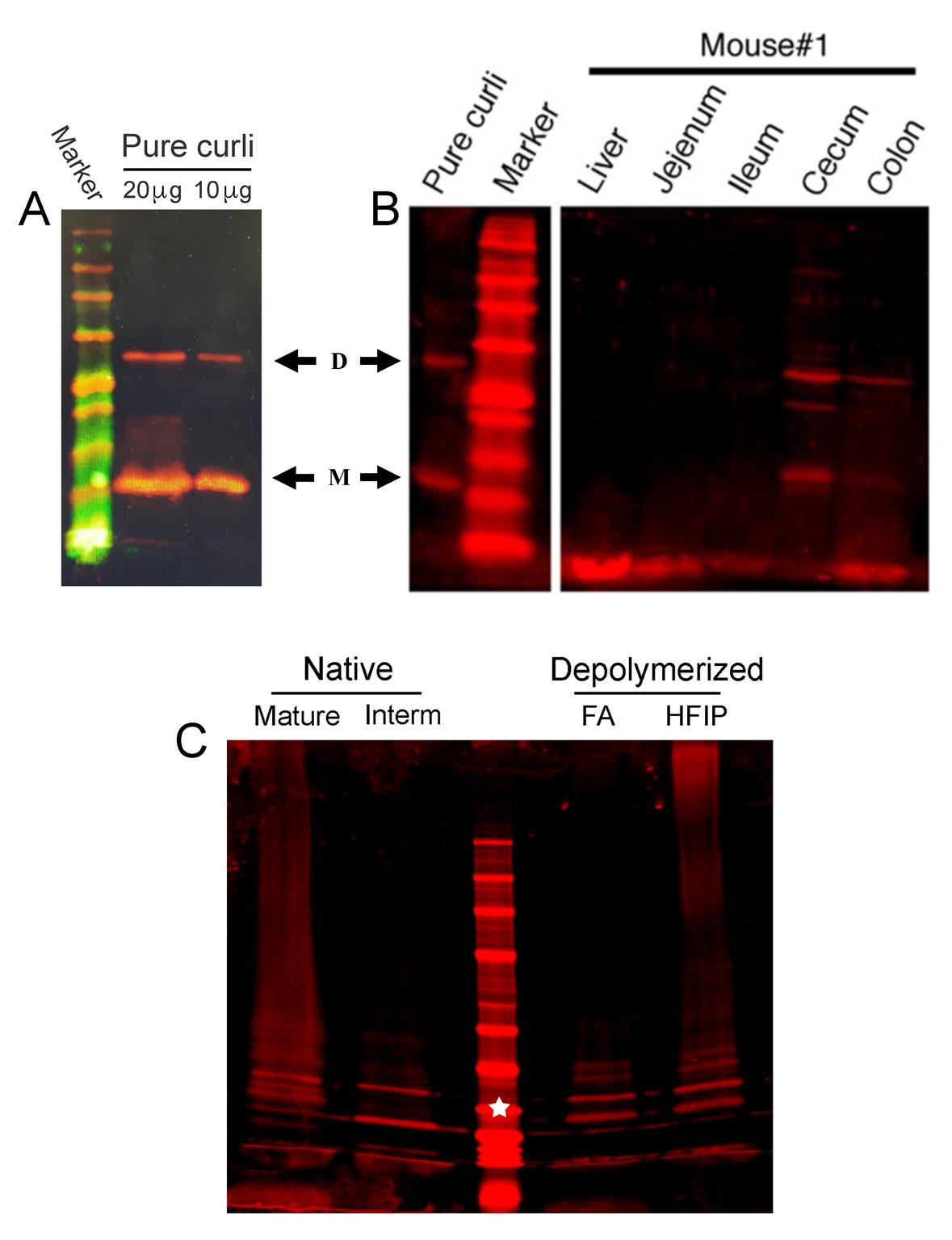
Figure 6. Comparison of different forms of curli as detected by immunoblot analysis. (A and B) Samples were resolved by SDS-PAGE consisting of a 5% acrylamide stacking gel and 12% acrylamide resolving gel. Proteins were transferred to nitrocellulose membranes using the iBlot system. For all immunoblots, monomer (M), dimer (D), and higher molecular weight oligomers of CsgA were detected using rabbit anti-curli polyclonal serum, followed by IRDye 680RD goat anti-rabbit IgG and detection using the Odyssey CLx imaging system (Li-Cor Biosciences). (A) 20 µg and 10 µg of purified curli were depolymerized with 90% FA prior to SDS-PAGE. (B) 50 mg samples of homogenized tissues from mice infected with S. Typhimurium were treated three successive times with 90% FA prior to SDS-PAGE, as previously described (Miller et al., 2020). (C) 10 µg of purified full-length curli (Mature), curli intermediates (Interm) and FA or HFIP treated curli were loaded and resolved directly on pre-cast NovexTM 4–12% Tris-Glycine Mini Gels. The white star denotes the 25 kDa protein standard.Purification of Curli Intermediates
Purification of curli intermediates relies on generating a ∆msbB mutant strain (Nicastro et al., 2019).
Studies on human amyloids have proved that different types of intermediate structures formed during the multistep process of amyloid polymerization. Wherein, soluble human amyloid monomers first form oligomers, which then polymerize into protofibrillar structures and then cross-assemble them into a stable, mature fibrils. Even though the process of curli fibrillar assembly in Enterobacteriaceae has been well-studied, the intermediate oligomeric structures of curli have not been identified or studied until recently by Nicastro et al. (2019). The authors have used the ∆msbB strain to understand the fibrillization kinetics of curli, their intermediates, as well as the role of mature curli fibrillar aggregates in the assembly of bacterial extracellular Matrix. The ∆msbB gene in Salmonella encodes the enzyme that catalyzes one of the two secondary acylation reactions that completes lipid A biosynthesis. It synthesizes a full-length O-antigen-containing LPS molecule that lacks only the expected secondary acyl chain, and is less able to induce cytokine and inducible nitric oxide synthase responses in both in vitro and in vivo conditions. They found that intermediate protofibrillar structures of bacterial amyloid (Curli intermediates) are more cytotoxic, and the addition of bacterial DNA accelerates them to form a mature fibrillar structure, limiting cytotoxic effects. The more detailed information regarding the kinetics of curli fibrilization and its stability can be found elsewhere (Nicastro et al., 2019). We used the lambda red recombinase knockout procedure (Datsenko and Wanner, 2000) to generate S. Typhimurium 14028s ∆msbB. Our first attempts to generate the ∆msbB strain were unsuccessful because we had difficulties recovering the antibiotic resistant transformants on LB agar. It has been reported that a Salmonella msbB mutant strain shows poor growth on LB agar and that growth can be restored to near wild-type levels by switching to MSB agar (LB with no NaCl supplemented with Mg2+ and Ca2+) (Murray et al., 2001). Once the procedure was changed to recover CMR (Chloramphenicol-resistant) transformants on MSB agar instead of LB agar, the lambda red procedure worked efficiently.
Grow strain S. Typhimurium 14028s ∆msbB in 5 mL of MSB broth with 34 µg/mL chloramphenicol overnight at 37°C with shaking at 200 rpm.
Add 5 mL of overnight culture to 500 mL of YESCA broth supplemented with 4% DMSO in a 1-L flask and incubate at 26°C for 72 h with shaking at 200 rpm.
After incubation, collect bacterial pellet by centrifugation at 10,000 × g for 10 min.
Resuspend the pellet in 30 mL of 10 mM Tris-HCl (pH 8.0) and treat with RNase A (0.1 mg/mL final conc.), DNase I (0.1 mg/mL final conc.), and MgCl2 (1 mM final conc.) for 30 min at 37°C.
After enzyme treatment, sonicate the suspension to break open the bacteria with 3 × 30 s pulses at 30% amplitude with 1 min cooling on ice between pulses.
To the mixture, add lysozyme to a final concentration of 1 mg/mL and incubate for 40 min at 37°C.
Add SDS to a final concentration of 1% to the mixture and incubate for 20 min at 37°C with shaking at 200 rpm.
After incubation, pellet curli by centrifugation (10,000 × g for 10 min at 4°C).
Resuspend the curli pellet in 10 mL of 10 mM Tris-HCl (pH 8.0) and boil for 10 min.
Cool down the sample on ice before adding RNase A (0.1 mg/mL final conc.), DNase I (0.1 mg/mL final conc.), lysozyme (1 mg/mL final conc.) and MgCl2 (1 mM final conc.). Incubate this mixture at 37°C for 2 h. Repeat this step until the desired viscosity is reached (see section A steps 13 and 14).
Centrifuge the mixture at 10,000 × g and 4°C for 10 min.
Wash the pellet three times with 10 mL 10 mM Tris-HCl (pH 8.0) and resuspend in 3 mL of 2× SDS-PAGE sample buffer and boil for 10 min.
Load the 3 mL of crude protein sample (from previous step) into a 4 mm-thick SDS-PAGE gel with 5% stacking gel and 12% separating gel and run continuously at 100 V until all the dye in the sample has run through the bottom.
After electrophoresis, use a 5 mL syringe with 18-gauge needle to collect the curli aggregates from the top of the gel, resuspend in 5–10 mL of sterile water, pellet by centrifugation (10,000 × g for 10 min) and wash two times with sterile water.
Resuspend the curli in 5 mL of 95% ethanol, pellet by centrifugation at 13,500 × g for 10 min, and wash two more times with ethanol.
Resuspend the curli in sterile water and transfer the mixture into a pre-weighed glass vial.
Freeze the mixture at -80°C and lyophilize for 24 h.
After lyophilization, weigh the glass vial containing sample to obtain the rough sample weight and store the lyophilized material at -20°C. The purity of curli intermediates can be checked by immunoblot analysis (Figure 6C).
Curli detection in host tissues/samples
The procedure for analysis of mouse tissues was adapted from Miller et al. (2020). The same procedure could be used to analyze tissues from other animal species or for other types of samples.
Place tissue samples (i.e., liver, cecum, colon, and small intestine) in tinfoil packets, snap freeze in liquid nitrogen, and store at -80°C.
Pre-chill a mortar and pestle by storing at -20°C. Grind the frozen tissue samples into a fine powder under liquid nitrogen. The powdered samples can be scraped out of the mortar and pestle using a metal spatula onto weigh paper and transferred in 2 mL screwcap freezer vials, and stored at -80°C.
Weigh 50 mg samples of powdered tissue, transfer into Eppendorf tubes and resuspend in 500 μL of 1× SDS-PAGE sample buffer and boil for 10 min. This step will dissolve most of the soluble cellular proteins.
Cool down the solution on ice, and pellet the cell debris by centrifugation at 25,000 × g for 5 min.
Resuspend the pellet in 500 μL of sterile distilled water and centrifuge at 25,000 × g for 5 min. Repeat this step twice. This washes away any remaining residues of SDS-PAGE sample buffer.
Resuspend the pellet with 500 μL of 90% FA and freeze the sample at -80°C for at least 1 h.
Lyophilize the mixture for 16–24 h. Repeat steps 6 and 7 three times. For purified curli standards, FA treatment only needs to be performed once.
Note: Samples containing acid may damage some lyophilizers.
After FA treatment, resuspend the lyophilized samples in 50 μL of 1× SDS-PAGE sample buffer and centrifuge the samples at 25,000 × g for 2 min.
Note: Do not boil the samples, because it will cause the curli monomers to aggregate.
Load a 20 μL aliquot of supernatant into each SDS-PAGE gel lane. We have used self-prepared 5% acrylamide stacking and 12% acrylamide resolving gels or commercial 4–12% acrylamide gradient gels. Load at least one gel lane with a commercial prestained protein ladder.
Perform electrophoresis at 110 V. Carefully remove and place the gel on the nitrocellulose membrane and perform protein transfer for 40 min at 25V in Trans-Blot SD semi-dry transfer cell. Alternatively, proteins can be transferred to nitrocellulose membranes using the automated iBlot system.
Detect curli proteins present in the tissue samples using any standard immunoblotting procedure. We commonly use rabbit anti-curli polyclonal serum as the primary antibody at 1:500 dilution in 5% skim milk in TBST and incubate at 4°C overnight or at 37°C for one hour. The secondary antibody used is either IRDye 680RD goat anti-rabbit IgG at 1:10,000 dilution (for fluorescent applications) or Goat-anti-rabbit IgG alkaline phosphatase conjugate at 1:2,000 dilution in 5% skim milk in TBST. The incubation is for 1 h 30 min at room temperature in the dark for fluorescence or 1 h at 37°C on a tilted platform shaker for colorimetric detection.
Antibody binding is visualized using fluorescence with the Odyssey CLX imaging system and Image Studio 4.0 software package (Figure 6B). For colorimetric detection of curli, soluble BCIP/NBT are used as substrates. Alkaline phosphatase will produce a stable blue-purple product that will not fade upon exposure to light, and you do not need special equipment to visualize.
DNA extraction from curli
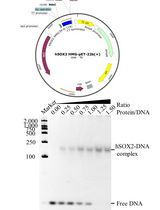
Previous work from the Tükel lab has shown that extracellular DNA (eDNA) associates with curli fibrils during the development of the mature biofilm and purified curli contains eDNA (Gallo et al., 2015). Since the presence of eDNA can influence immune recognition as well as TLR2/9 stimulation (Tursi and Tükel, 2018), it is good practice to determine just how much eDNA is associated with purified curli each time a batch is purified.
Resuspend 500 μg of purified curli in 550 μL of TE buffer, 30 μL of 10% SDS, and 70 μL of proteinase K (20 mg/mL).
Mix thoroughly and incubate at 37°C for 1 h.
Add 100 μL of 5 M NaCl and mix by pipetting.
Add 80 μL of CTAB/NaCl solution and mix thoroughly by pipetting using wide bore tips.
Incubate at 37°C for 10 min.
Add 300–400 μL phenol/chloroform/isoamyl alcohol to samples and mix by inverting the tubes
Centrifuge at 15,682 × g and 4°C for 5 min.
Transfer upper phase to a new Eppendorf tube.
Add 700 μL of chloroform and mix by pipetting.
Centrifuge at 15,682 × g and 4°C for 5 min.
Transfer upper phase to a new Eppendorf tube and add equal volume isopropanol and shake the tube by hand.
Incubate at -20°C for 30 min.
Centrifuge at 13,362 × g and 4°C for 5 min.
Discard the liquid and rinse the DNA pellet with 1 mL of 70% EtOH and centrifuge for 5 min at 5,220 × g.
Resuspend the final DNA pellet in 30 μL of TE buffer or water.
Measure the DNA content using a DeNovix DS-11 FX spectrophotomer/fluorometer.
Data analysis
For detection of bacterial produced curli from animal tissues, it is recommended to test at least two 50 mg samples from each tissue type, and to screen all individual mice from infected groups. In our previous work, we did not detect the presence of curli in each mouse that was infected with Salmonella (Miller et al., 2020). We are unsure if this was because of stochastic expression of curli (i.e., it is not produced in each mouse) or due to technical difficulties with detection. For this reason, the authors recommend that each individual animal within a group be tested.
Recipes
MSB broth (1,000 mL)
Tryptone 10 g
Yeast Extract 2.5 g
1 mL of 1 M CaCl2
1 mL of 1 M MgSO4
Autoclave the solution on a standard liquid cycle (20 min at 15 psi).
YESCA broth (1,000 mL)
Casamino acid 10 g
Yeast Extract 1 g
Autoclave the solution on a standard liquid cycle (20 min at 15 psi) and add DMSO (final concentration 4%).
T Agar (1,000 mL)
Tryptone 10 g
Agar 15 g
Autoclave the solution on a standard liquid cycle (20 min at 15 psi).
2× SDS-PAGE sample buffer (100 mL)
Tris (1 M, pH 6.8) 8 mL
SDS (20%) 10 mL
Glycerol 10 mL
Bromophenol blue (0.1%) 600 μL
β-mercaptoethanol (add fresh) 4 mL
Distilled water 67.4 mL
10× SDS-PAGE running buffer (1,000 mL)
Tris base 30.3 g
Glycine 144.4 g
SDS 10 g
10× Towbin buffer or Transfer buffer (1,000 mL)
Tris base 30.3 g
Glycine 144.4 g
Prepare 1× transfer buffer by adding 100 mL of 10× transfer buffer with 200 mL of methanol and add 700 mL of distilled water.
10× TBS (1,000 mL)
Tris base 24 g
NaCl 88 g
Dissolve the components in 900 mL of distilled water. Adjust the pH to 7.6 with HCl and bring up the final volume with distilled water. Prepare 1× TBS by adding 100 mL of 10× TBS with 900 mL of distilled water.
1× TBST
Add 0.5 mL of Tween 20 to 1,000 mL of 1× TBS.
12% separating gel (2 gels)
Distilled water 82.56 mL
1.5 M Tris-HCl pH 8.8 48 mL
Acrylamide/bis (40%) 57.6 mL
10% SDS 1.92 mL
10% APS 1.92 mL
TEMED 96 μL
5% stacking gel (2 gels)
Distilled water 23.84 mL
0.5 M Tris-HCl pH 6.8 9.6 mL
Acrylamide/bis (40%) 4.5 mL
10% SDS 400 μL
10% APS 480 μL
TEMED 40 μL
AP buffer (500 mL)
100 mM Tris 6.06 g
100 mM NaCl 2.92 g
5 mM MgCl2·6H2O 0.51 g
Dissolve the components in 450 mL of distilled water. Adjust the pH to 9.5 with 3 M HCl and bring up to 500 mL with distilled water and autoclave.
NBT/BCIP developing solution
AP buffer 10 mL
NBT 66 μL
BCIP 33 μL
Acknowledgments
This research was supported by a Natural Sciences and Engineering Research Council (NSERC) Discovery grant (Grant #2017-05737), the Jarislowsky Chair in Biotechnology, and a College of Medicine Research Award from the University of Saskatchewan to A.P.W. CT was supported by NIH grants AI153325, AI151893, and AI148770. E.G.H., was supported by an NSERC undergraduate research award. We thank Neil Rawlyk at VIDO for general technical assistance. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The mature curli purification protocol and immunodetection of curli from animal tissues are used in our recent publication (Miller et al., 2020). The protocols for purification of curli intermediates and DNA extraction were published in Nicastro et al. (2019). Published as VIDO manuscript series No. 968.
Competing interests
The authors have no competing interests to report.
Ethics
Mice were cared for and used in accordance with the Guidelines of the Canadian Council on Animal Care and the Regulations of the University of Saskatchewan Committee on Animal Care and Supply, following Animal Use Protocols #20110057 or 20170080, which were approved by the University of Saskatchewan’s Animal Research Ethics Board.
References
- Anriany, Y. A., Weiner, R. M., Johnson, J. A., De Rezende, C. E. and Joseph, S. W. (2001). Salmonella enterica serovar Typhimurium DT104 displays a rugose phenotype. Appl Environ Microbiol 67(9): 4048-4056.
- Bian, Z., Brauner, A., Li, Y. and Normark, S. (2000). Expression of and cytokine activation by Escherichia coli curi fibers in human sepsis. J Infect Dis 181(2): 602-612.
- Brombacher, E., Baratto, A., Dorel, C. and Landini, P. (2006). Gene expression regulation by the Curli activator CsgD protein: modulation of cellulose biosynthesis and control of negative determinants for microbial adhesion. J Bacteriol 188(6): 2027–2037.
- Chapman, M.R., Robinson, L.S., Pinkner, J.S., Roth, R., Heuser, J., Hammar, M., Normark, S. and Hultgren, S.J. (2002). Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295(5556): 851-855.
- Collinson, S. K., Emödy, L., Müller, K. H., Trust, T. J. and Kay, W. W. (1991). Purification and characterization of thin, aggregative fimbriae from Salmonella enteritidis. J Bacteriol 173(15): 4773-4781.
- Collinson, S. K., Parker, J. M. R., Hodges, R. S. and Kay, W. W. (1999). Structural predictions of AgfA, the insoluble fimbrial subunit of Salmonella thin aggregative fimbriae. J Mol Biol 290(3): 741-756.
- Datsenko, K.A. and Wanner, B.L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97: 6640-6645.
- Evans, M. L. and Chapman, M. R. (2014). Curli biogenesis: order out of disorder. Biochim Biophys Acta Mol Cell Res 1843(8): 1551-1558.
- Gallo, P. M., Rapsinski, G. J., Wilson, R. P., Oppong, G. O., Sriram, U., Goulian, M., Buttaro, B., Caricchio, R., Gallucci, S. and Tukel, C. (2015). Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity 42(6): 1171-1184.
- Gerstel, U. and Römling, U. (2003). The csgD promoter, a control unit for biofilm formation in Salmonella typhimurium. Res Microbiol 154(10):659-67.
- Miller, A. L., Pasternak, J. A., Medeiros, N. J., Nicastro, L. K., Tursi, S. A., Hansen, E. G., Krochak, R., Sokaribo, A. S., MacKenzie, K. D., Palmer, M. B., et al. (2020). In vivo synthesis of bacterial amyloid curli contributes to joint inflammation during S. Typhimurium infection. PLoS Pathog 16(7): e1008591.
- Müller, K. H., Collinson, S. K., Trust, T. J. and Kay, W. W. (1991). Type 1 fimbriae of Salmonella enteritidis. J Bacteriol 173(15): 4765-4772.
- Murray, S. R., Bermudes, D., de Felipe, K. S. and Low, K. B. (2001). Extragenic suppressors of growth defects in msbB Salmonella. J Bacteriol 183(19): 5554-5561.
- Nicastro, L. K., Tursi, S. A., Le, L. S., Miller, A. L., Efimov, A., Buttaro, B., Tam, V. and Tukel, C. (2019). Cytotoxic Curli Intermediates Form during Salmonella Biofilm Development. J Bacteriol 201(18): e00095-19.
- Olsén, A., Wick, M.J., Morgelin, M. and Bjorck, L. (1998). Curli, fibrous surface proteins of Escherichia coli, interact with major histocompatibility complex class I molecules. Infect Immun 66(3): 944-949.
- Olsén, A., Jonsson, A. and Normark, S. (1989). Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli. Nature 338(6217): 652-655.
- Perov, S., Lidor, O., Salinas, N., Golan, N., Tayeb-Fligelman, E., Deshmukh, M., Willbold, D. and Landau, M. (2019). Structural insights into curli CsgA cross-β fibril architecture inspire repurposing of anti-amyloid compounds as anti-biofilm agents. PLoS Pathog 15(8): e1007978.
- Rapsinski, G. J., Wynosky-Dolfi, M. A., Oppong, G. O., Tursi, S. A., Wilson, R. P., Brodsky, I. E. and Tükel, Ç. (2015). Toll-like receptor 2 and NLRP3 cooperate to recognize a functional bacterial amyloid, curli. Infect Immun 83(2): 693-701.
- Römling, U., Bian, Z., Hammar, M., Sierralta, W. D. and Normark, S. (1998). Curli fibers are highly conserved between Salmonella typhimurium and Escherichia coli with respect to operon structure and regulation.J Bacteriol 180(3): 722-731.
- Ryu, J. H. and Beuchat, L. R. (2005). Biofilm formation by Escherichia coli O157: H7 on stainless steel: effect of exopolysaccharide and curli production on its resistance to chlorine. Appl Environ Microbiol 71(1): 247-254.
- Scher, K., Romling, U. and Yaron, S. (2005). Effect of heat, acidification, and chlorination on Salmonella enterica serovar typhimurium cells in a biofilm formed at the air-liquid interface. Appl Environ Microbiol 71:1163-1168.
- Sjobring, U., Pohl, G. and Olsen, A. (1994). Plasminogen, absorbed by Escherichia coli expressing curli or by Salmonella enteritidis expressing thin aggregative fimbriae, can be activated by simultaneously captured tis- sue-type plasminogen activator (t-PA). Mol Microbiol 14(3):443-452.
- Solano, C., García, B., Valle, J., Berasain, C., Ghigo, J. M., Gamazo, C. and Lasa, I. (2002). Genetic analysis of Salmonella enteritidis biofilm formation: critical role of cellulose. Mol Microbiol 43(3): 793-808.
- Szulc, N., Gąsior-Głogowska, M., Wojciechowski, J. W., Szefczyk, M., Żak, A. M., Burdukiewicz, M. and Kotulska, M. (2021). Variability of Amyloid Propensity in Imperfect Repeats of CsgA Protein of Salmonella enterica and Escherichia coli. Int J Mol Sci 22(10): 5127.
- Tükel, Ç., Nishimori, J. H., Wilson, R. P., Winter, M. G., Keestra, A. M., Van Putten, J. P. and Bäumler, A. J. (2010). Toll‐like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms. Cell Microbiol 12(10): 1495-1505.
- Tükel, Ç., Raffatellu, M., Humphries, A. D., Wilson, R. P., Andrews‐Polymenis, H. L., Gull, T., Figueiredo, J. F., Wong, M. H., Michelsen, K. S., Akçelik, M., Adams, L. G., et al. (2005). CsgA is a pathogen‐associated molecular pattern of Salmonella enterica serotype Typhimurium that is recognized by Toll‐like receptor 2. Mol Microbiol 58(1): 289-304.
- Tursi, S. A. and Tükel, Ç. (2018). Curli-containing enteric biofilms inside and out: matrix composition, immune recognition, and disease implications. Microbiol Mol Biol Rev 82(4): e00028-18.
- White, A. P., Gibson, D. L., Collinson, S. K., Banser, P. A. and Kay, W. W. (2003). Extracellular polysaccharides associated with thin aggregative fimbriae of Salmonella enterica serovar Enteritidis. J Bacteriol, 185(18), 5398-5407.
- White, A. P., Gibson, D. L., Kim, W., Kay, W. W. and Surette, M. G. (2006). Thin aggregative fimbriae and cellulose enhance long-term survival and persistence of Salmonella. J Bacteriol 188(9): 3219-3227.
- White, A. P. and Surette, M. G. (2006). Comparative genetics of the rdar morphotype in Salmonella. J Bacteriol 188(24): 8395-8406.
- Zhou, Y., Smith, D. R., Hufnagel, D. A. and Chapman, M. R. (2013). Experimental manipulation of the microbial functional amyloid called curli. Methods Mol Biol 966: 53-75.
- Zogaj, X., Nimtz, M., Rohde, M., Bokranz, W. and Römling, U. (2001). The multicellular morphotypes of Salmonella typhimurium and Escherichia coli produce cellulose as the second component of the extracellular matrix. Mol Microbiol 39(6): 1452-1463.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sivaranjani, M., Hansen, E. G., Perera, S. R., Flores, P. A., Tukel, C. and White, A. P. (2022). Purification of the Bacterial Amyloid “Curli” from Salmonella enterica Serovar Typhimurium and Detection of Curli from Infected Host Tissues. Bio-protocol 12(10): e4419. DOI: 10.21769/BioProtoc.4419.
Category
Microbiology > Microbe-host interactions > Bacterium
Microbiology > Microbial biofilm > Biofilm culture
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.