- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Rapid Generation of Human Neuronal Cell Models Enabling Inducible Expression of Proteins-of-interest for Functional Studies
Published: Vol 10, Iss 9, May 5, 2020 DOI: 10.21769/BioProtoc.3615 Views: 5318
Reviewed by: Ehsan KheradpezhouhNarayan SubramanianAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
CRISPR-Cas9 technology has transformed the ability to edit genomic sequences and control gene expression with unprecedented ease and scale. However, precise genomic insertions of coding sequences using this technology remain time-consuming and inefficient because they require introducing adjacent single-strand cuts through Cas9 nickase action and invoking the host-encoded homology-directed repair program through the concomitant introduction of large repair templates. Here, we present a system for the rapid study of any protein-of-interest in two neuronal cell models following its inducible expression from the human AAVS1 safe harbor locus. With lox-flanked foundation cassettes in the AAVS1 site and a tailor-made plasmid for accepting coding sequences-of-interest in place, the system allows investigators to produce their own neuronal cell models for the inducible expression of any coding sequence in less than a month. Due to the availability of preinserted enhanced green fluorescent protein (EGFP) coding sequences that can be fused to the protein-of-interest, the system facilitates functional investigations that track a protein-of-interest by live-cell microscopy as well as interactome analyses that capitalize on the availability of exquisitely efficient EGFP capture matrices.
Keywords: Human neuronal cellsBackground
The ability to engineer the genomes of cell models and organisms holds tremendous potential for research and targeted therapy. With the advent of CRISPR-Cas9 technology, the efficiency of precision genome engineering has greatly improved, and useful reagents are coming online at a rapid pace. Despite these advances, there still is a scarcity of cell-based in vitro paradigms for studying neurological diseases that is holding back the pace of progress in this area. Whereas CRISPR-Cas9 mediated homology-directed repair (HDR) can now easily be applied to generate knockout models or to knock in small transgenic segments, the goal to integrate larger segments continues to present a considerable challenge (Devkota, 2018).
In our own work, we often would like to learn more about the function of a protein by studying its subcellular localization and protein-protein interactions. To this end, it would help if neuronal cell models could be equipped with inducible expression systems that provide temporal control of the expression of a protein-of-interest and facilitate its visualization and capture. Not long ago, this need would call for the stable transfection or lentiviral transduction of an expression cassette, an approach that can result in unpredictable confounding effects due to the random integration of transgenes. CRISPR-Cas9 provides alternative strategies to accomplish this task in a precise way but these approaches remain time-consuming and require considerable investments in resources and expertise.
To address this unmet need, we sought to develop resources that enable the rapid and flexible integration of large inducible expression cassettes into the AAVS1 human safe harbor locus (DeKelver et al., 2010) of several neuronal cell models. Stable insertion into the AAVS1 locus, as opposed to viral integration, was chosen to preclude transgene silencing and insertional mutagenesis, notorious confounders associated with non-directed transgene insertions. To maximize flexibility and speed, the system was built around two genome-editing steps. The first step, which makes use of CRISPR-Cas9-mediated HDR followed by selection of positive clones, is time-consuming and uncertain to succeed but we have already accomplished this in 3 cell lines that are available to be shared. The second step, which relies on a Cre recombinase-mediated exchange of an expression cassette, is relatively easy to accomplish and fast because it can be followed by puromycin-based selection of positive clones.
More specifically, a paired Cas9 nickase CRISPR strategy, used to improve the specificity of targeting and to minimize off-target effects (Ran et al., 2013), was employed to insert a foundation cassette (FC), comprising a pair of lox sites flanking a G418 resistance marker into Intron 1 of the AAVS1 locus, a human genetic safe harbour. In parallel, a ~7 kb inducible expression plasmid (IEX) was built. The expression cassette within the IEX is flanked by compatible lox sites and comprises elements designed to accept the insertion and control the expression of any coding sequence-of-interest (Figure 1). More specifically, the IEX comprises a promoter-less puromycin resistance marker, the reverse transactivator (rtTA3), and an EGFP tag fused to a protein-of-interest insertion site. With this arrangement, the rtTA3 is designed to drive the inducible expression of the EGFP-fused protein-of-interest that is under the control of the TREtight inducible promoter.
With these tools in hand, novel cell models for the inducible expression of a protein-of-interest fused to EGFP can be generated in three straightforward steps:
1.Insert coding sequence for protein-of-interest into the IEX plasmid.
2.Transiently co-transfect the IEX plasmid and a plasmid coding for Cre recombinase into the parental cell equipped with one or two FCs.
3.Select stable integrant clones by puromycin selection or fluorescence activated cell sorting (FACS).
In addition to its ease of handling and speed, the system restricts the copy number of insertions to two, corresponding to the two AAVS1 alleles present in the human genome. The system also lends itself to comparative analyses of several proteins, because its design ensures that cells coding for different proteins-of-interest can be generated side-by-side, requiring no specific resources, except for a human cDNA library and the corresponding primers to amplify and insert a given coding sequence into the IEX cassette. Note that fusing the coding sequence for a protein-of-interest to EGFP does not only provide the ability to undertake live cell imaging but the EGFP tag also allows the high affinity purification of fusion proteins by GFP nanotrap technology for the study of protein-protein interactions (Rothbauer et al., 2008).
We recently applied the system to study the TAU protein and BAG5 in several human neuronal cell models, including ReN VM human neural progenitor cells that can be differentiated into a co-culture of neural and glial cells (Donato et al., 2007). These studies have led us to detect subtle differences in protein-protein interactions of wild-type versus mutant forms of TAU and BAG5 (Figure 1). This article was written with the intent to share these resources and facilitate their adaptation to the study of other proteins-of-interest.
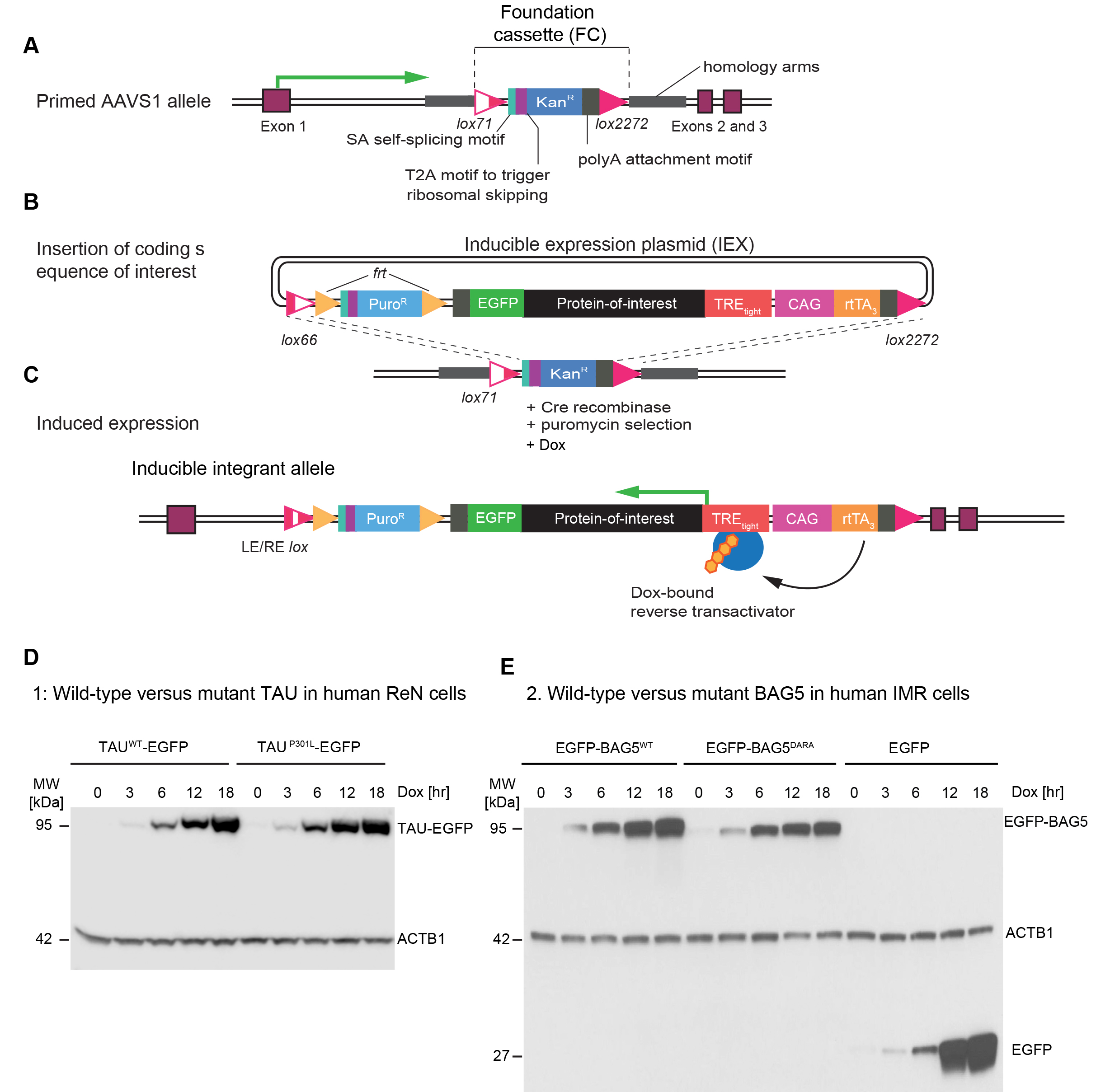
Figure 1. Design of AAVS1 embedded system for inducible expression of proteins-of-interest. A. CRISPR-Cas9 nickase-mediated insertion of foundation cassette (FC) into AAVS1 safe harbour. B. Insertion of coding sequence-of-interest into inducible expression plasmid (IEX). C. Reverse-tetracycline-controlled transactivator (rtTA3)-mediated expression of protein-of-interest following addition of doxycycline to cell culture medium. D. Example 1: Parallel induction of wild-type and P301L mutant TAU fused to C-terminal EGFP in ReN cells. Antibodies directed against beta-actin served as the loading control. E. Example 2: Side-by-side induction of N-terminally EGFP-tagged wild-type or mutant (DARA) BAG5. Cells expressing EGFP only served as additional controls in this analysis.
Materials and Reagents
- Biological materials
- Plasmids (Table 1)
- Cell lines (Table 2)
Table 1. List of available plasmids for generating inducible cells and their Addgene ID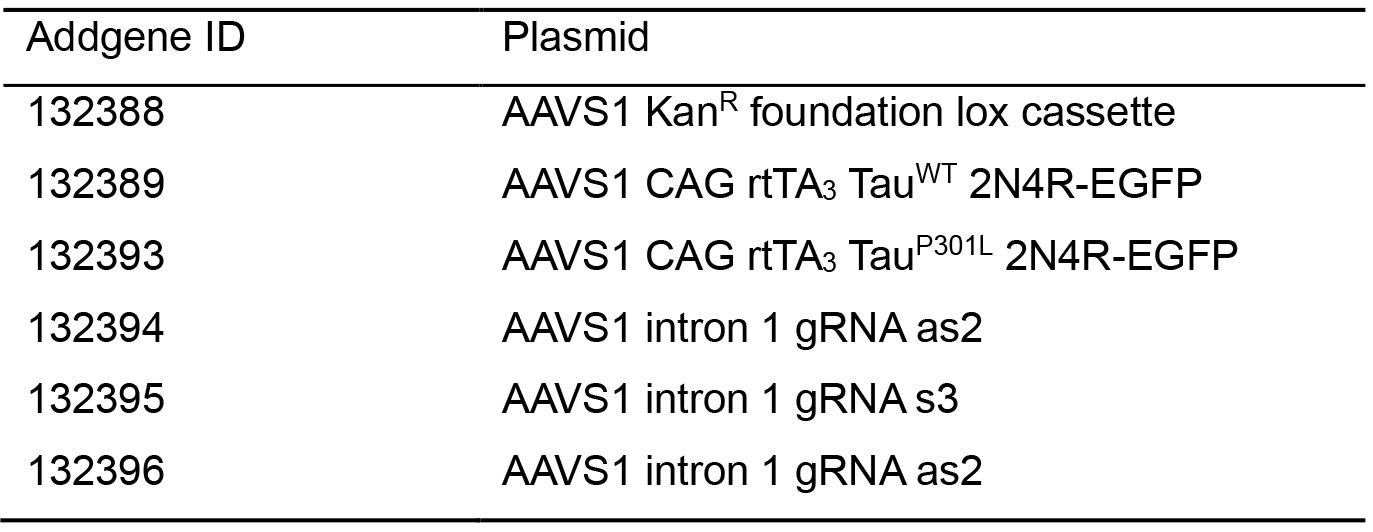
Table 2. List of available cell lines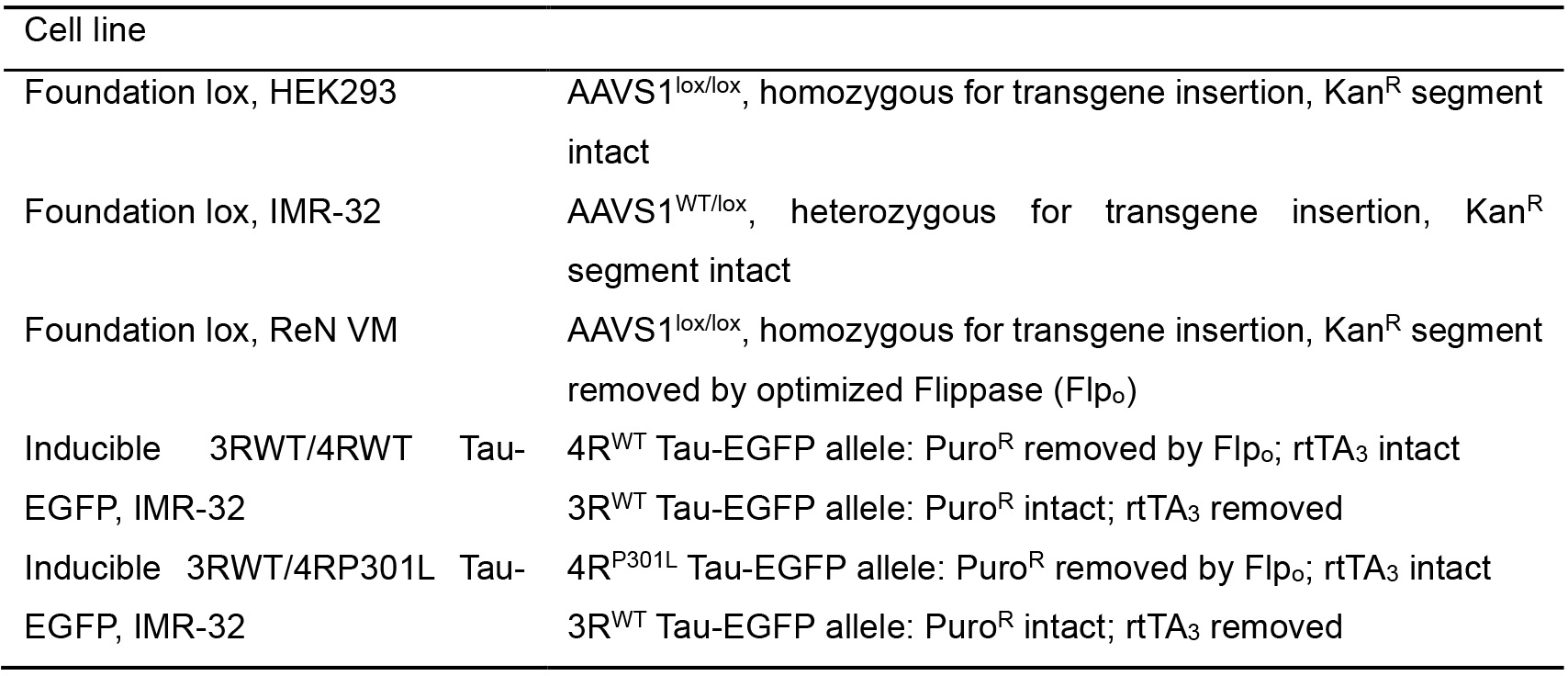
- Cloning
Note: All cloning enzymes and PCR reagents are stored at -20 °C, stable for 1 year.- Q5® Hot Start high-fidelity 2x master mix (New England Biolabs, catalog number: M0494S )
- Primers: ordered as Value Oligos or Custom Oligos, 25 nmole, desalted (Invitrogen). Reconstitute to 100 μM stocks in water, store at -20 °C
- Q5® Site-directed mutagenesis kit (New England Biolabs, catalog number: E0554S )
- EZ-10 spin column PCR products purification kit (BioBasic, catalog number: BS664-250preps )
- Restriction enzyme BbvCI with CutSmart buffer (New England Biolabs, catalog number: R0601S )
- Restriction enzyme NheI-HF with CutSmart buffer (New England Biolabs, CutSmart, catalog number: R3131S )
- Fast AP thermosensitive alkaline phosphatase (Thermo Fisher Scientific, catalog number: EF0651 )
- PureLink Quick Gel Extraction Kit (Invitrogen, catalog number: K210012 )
- T4 DNA ligase (New England Biolabs, catalog number: M0202S )
- EnduraTM Chemically Competent E. coli Cells (Lucigen, catalog number: 60241-2 ). Store at
-80 °C - EZ-10 spin column plasmid DNA minipreps kit (BioBasic, catalog number: BS614-250preps )
- PureLink® HiPure plasmid filter maxi kit (Invitrogen, catalog number: K210026 )
- Ampicillin sodium salt powder, 91.0-100.5%, cell culture tested (Sigma-Aldrich, catalog number: A1066-5G ). Store at 4 °C, stable for 1 year
- Inducible cell generation
- paavCAG-iCre plasmid (Addgene, catalog number: 51904 , a gift from Jinhyn Kim)
- jetPRIME (PolyPlus, catalog number: 114-07 ). Store at 4 °C, stable for 1 year
- TransfeX transfection reagent (ATCC, catalog number: ACS-4005 ). Store at 4 °C, stable for 1 year
- Millex-HA syringe filters, 0.45 μm pore size (Millipore, catalog number: 5010-SLHA033SS )
- Millex-GP syringe filters, 0.22 μm pore size (Millipore, catalog number: SLGP033RS )
- 6-well tissue-culture plates (Falcon, catalog number: 353046 )
- 24-well tissue-culture plates (Falcon, catalog number: 353047 )
- Puromycin (Sigma-Aldrich, catalog number: P7255-25MG ). Reconstitute to 10 mg/ml stock, filter sterilize, aliquot, store at -20 °C
- Doxycycline (BioBasic, catalog number: DB0889-25G ). Reconstitute to 10 mg/ml stock, aliquot, store at -20 °C
- Affinity capture with GFP nanotrap
- Cell culture
- 6-cm tissue-culture plates (Sarstedt, catalog number: 83.3901 )
- 1.5 ml centrifuge tubes (Sarstedt, catalog number: 72.690.300 )
- 12-well tissue-culture plates (Sarstedt, catalog number: 83.3921 )
- Dulbecco's Phosphate-Buffered Saline (D-PBS), without calcium chloride and magnesium chloride, liquidPhosphate buffered saline (Sigma-Aldrich, catalog number: D8537-500ML ). Store at 4 °C, stable for 1 year5.
- 0.25% Trypsin with EDTA 4Na (Gibco, catalog number: 25200072 ). Aliquot, store at -20 °C for long-term storage. Store at 4 °C for short-term use, stable for 6 months
- StemPro® Accutase® Cell Dissociation Reagent (Gibco, catalog number: A1110501 ). Aliquot, store at -20 °C for long-term storage. Store at 4 °C for short-term use, stable for 6 months
- Matrigel, growth-factor reduced (Corning, catalog number: 354230 ). Aliquot, store at -20 °C, stable for 1 year
- RecoveryTM Cell Culture Freezing Medium (Gibco, catalog number: 12648010 ). Aliquot, store at -20 °C, stable for 1 year
- DMEM with high glucose, L-glutamine, and sodium pyruvate (Gibco, catalog number: 11995073 ). Store at 4 °C, stable for 1 year
- 10% Fetal Bovine Serum, qualified (Gibco, catalog number: 12483020 ). Aliquot, store at -20 °C for long-term storage
- 1% GlutaMAXTM-I supplement (Gibco, catalog number: 35050061 ). Store at 4 °C, for stable for 1 year
- DMEM/F12, 1:1, contains L-glutamine, but no HEPES (Gibco, catalog number: 21041025 ). Store at 4 °C, stable for 1 year
- 2% N21-MAX supplement (R&D Systems, catalog number AR008 ) or 2% B-27 serum-free supplement (Gibco, catalog number: 17504044 ). Store at -20 °C, stable for 1 year
- Basic fibroblast growth factor (Gibco, catalog number: PHG0261 ). Store at -80 °C, stable for 1 year
- Epidermal growth factor (Reprokine, catalog number: RKP01133 ). Store at -80 °C, stable for 1 year
- Heparin (Sigma-Aldrich, catalog number: H3149-10KU ). Store at -80 °C, stable for 1 year
- 2 ng/ml glial-derived neurotrophic factor (Gibco, catalog number: PHC7045 )
- 500 μM dibutyryl-cyclic-adenosine monophosphate (Sigma-Aldrich, catalog number: D0627-250MG )
- Proliferation media for IMR-32 and HEK-293 cells (see Recipes)
- Proliferation media for ReN VM cells (see Recipes)
- Neuronal differentiation media for ReN VM cells (see Recipes)
- Cell lysis
Note: Original reagents are stored at room temperature. Reconstituted stocks are stored at 4 °C for 3 months. All buffers (see Recipes) are prepared fresh from reagent stocks immediately before use.- Tris-HCl, > 99% (BioShop, catalog number: TRS002.5 ). Reconstitute to 1 M stock in water
- NP-40 (BioShop, catalog number: NON505.500 ). Reconstitute to 10% stock in water
- Sodium deoxycholate (DOC), > 99% (BioShop, catalog number: DCA333.50 ). Reconstitute to 10% w/v stock in water, pH 8.0
- Sodium chloride (NaCl), > 99% (BioShop, catalog number: SOD002.205 ). Reconstitute to 5 M stock in water
- Ethylenediaminetetraacetate (EDTA), >99% (BioShop, catalog number: EDT001.500 ). Reconstitute to 0.5 M stock in water
- Sodium orthovanadate (Na3VO4) (BioShop, catalog number: SOV664.25 ). Reconstitute to 0.1 M stock in water
- Sodium fluoride (NaF) (BioShop, catalog number: SFL001.100 ). Reconstitute to 1 M stock in water
- Phenylmethylsulfonyl fluoride (PMSF) (BioShop, catalog number: PMS123.5 ). Reconstitute to 0.5 M stock in ethanol
- cOmplete protease inhibitor (Roche, catalog number: 11836170001 )
- PhosStop phosphatase inhibitor (Roche, catalog number: 4906837001 )
- HEPES (Bioshop, catalog number: HEP001.500 ). Reconstitute to 0.1 M stock in water
- Trifluoroacetic acid (TFA), > 99%, HPLC grade (Sigma-Aldrich, catalog number: 302031-100ML )
- Acetonitrile (ACN), HPLC grade (Caledon, catalog number: 1401-7-40 )
- Lysis buffer (see Recipes)
- Wash buffer (see Recipes)
- Elution buffer (see Recipes)
Equipment
- -80 °C freezer (Thermo Fisher, model: Standard Performance Ultra-Low Freezers )
- Refrigerated micro centrifuge (Eppendorf, model: 5424R , catalog number: 5404000138 )
- Avanti J-26S series high-speed centrifuge (Beckman-Coulter, catalog number: B22984 )
- Orbital shaker (Jeio Tech, model: ISF-7000 series )
- Fluorescence microscope (Leica Microsystems, model: Leica DMI 6000 B )
- NanoDrop spectrophotometer (Thermo Fischer Scientific, catalog number: ND-1000 )
- ESBE Scientific Save cell UV cell culture incubator (Panasonic, catalog number: MCO-19AIC )
- SterilGARD biosafety cabinet (The Baker Company, catalog number: SG603A-HE )
Software
- Snapgene (GSL Biotech LLC, https://www.snapgene.com/)
- Snapgene Viewer (GSL Biotech LLC, https://www.snapgene.com/snapgene-viewer/)
- Tm Calculator (New England Biolabs, https://tmcalculator.neb.com/#!/main)
- NEBioCalculator (New England Biolabs, https://nebiocalculator.neb.com/#!/ligation)
- NanoDrop 1000 (Thermo Fischer Scientific, https://www.thermofisher.com/ca/en/home/industrial/spectroscopy-elemental-isotope-analysis/molecular-spectroscopy/ultraviolet-visible-visible-spectrophotometry-uv-vis-vis/uv-vis-vis-instruments/nanodrop-microvolume-spectrophotometers/nanodrop-software-download.html)
Procedure
The protocol is designed to enable the generation of neural cell models for functional analyses of proteins-of-interest in less than a month (Figure 2). The system is scalable by parallelization of procedures.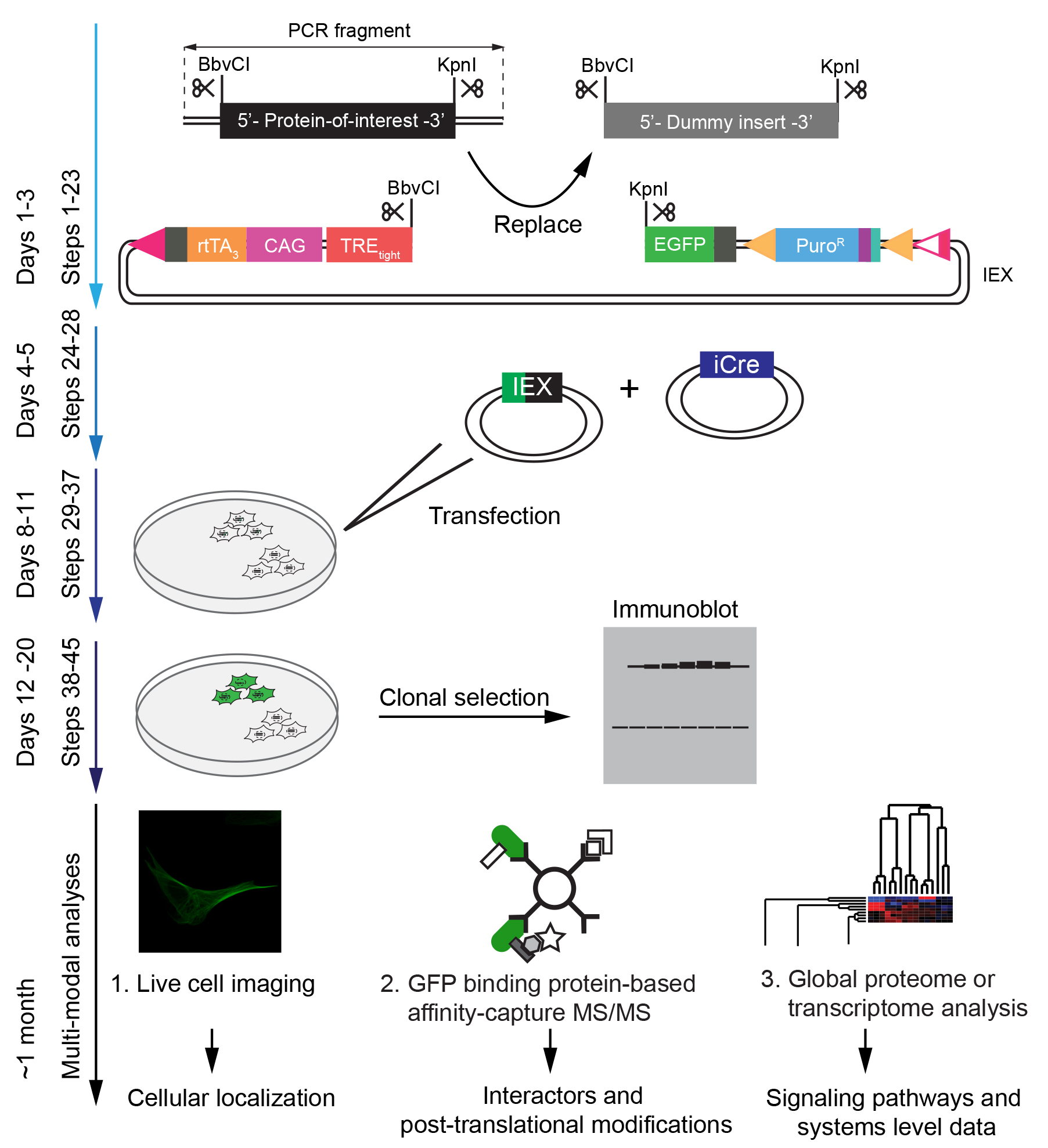
Figure 2. Time-line for the execution of procedures described in this protocol. All steps described in the protocol take approximately one month to implement. The generation of vectors coding for the inducible expression of a protein-of-interest can be completed in approximately one week. Once positive clones have been selected and validated, cell-based and biochemical analyses can be undertaken in parallel. Naturally, the one-month timeline will be exceeded, if complex follow-on downstream analyses are planned.
- Week 1. Cloning a gene of interest into the IEX
Notes:- Cloning strategies can be simulated, verified, and visualized using Snapgene and Snapgene Viewer.
- The coding sequence (CDS) of a new protein-of-interest can be cloned into the IEX using the unique restriction enzyme sites BbvCI and KpnI:
BbvCI: located 5’ of the start codon of new CDS.
KpnI: present in the linker region connecting EGFP to the C-terminus of the new protein. - Multiple other unique restriction enzyme sites (e.g., NheI, BstBI, KflI, etc.) may also be used.
- Order primers to append BbvCI and KpnI restriction enzyme sites to the 5’ and 3’ ends of the CDS of the protein-of-interest, respectively.
Example:Primer name Sequence Insert BbvCI site 5′…CCV TCAGC…3′ 5′ N8CCTCAGCGCCACCATGX20-25 3′ Insert KpnI site 5′…GGTAV CC…3′ 5′ N8GGTACCX20-25 3′ - N8 represent 8 random nucleotides to serve as overhangs required by restriction enzymes for efficient digestion.
- X20-25 represent sequences of the CDS of the protein-of-interest.
- Blue: Kozak sequence, including the ATG start codon, required for translation.
- Do not include a stop codon in the X20-25 sequence used for designing the ‘Insert KpnI’ primer if the new protein is to be expressed as an EGFP fusion protein.
- Determine the annealing temperature of primers using the New England Biolabs Tm Calculator tool, which returns this information when primer sequences are entered into the online form field at: https://tmcalculator.neb.com/#!/main.
- Perform PCR to append BbvCI and KpnI sites to CDS of protein-of-interest using the following PCR reaction:
Reagents 50 μl reaction (µl) Final concentration Q5 HiFi Hot start master mix (2x) 25 1x Template DNA (20 ng) Calculate ‘Insert BbvCI’ primer (10 μM) 2.5 0.5 µM ‘Insert KpnI’ primer (10 μM) 2.5 0.5 µM Nuclease-free H2O Add to 50 Total 50 - Perform the following PCR cycles:
Step Temperature (°C) Time Initial denaturation 98 30 s Denaturation 98 10 s 30 cycles Annealing To be determined 30 s Extension 72 10-20 s/kb Final extension 72 2 min Hold 4 Until sample storage - Proceed to PCR purification per manufacturer’s protocol. Measure DNA concentration on NanoDrop 1000 as per the manufacturer’s protocol.
- Set up restriction digest of IEX vector and new CDS insert using the following reactions:
Reagents IEX vector New CDS insert BbvCI 4 µl 4 µl KpnI 4 µl 4 µl CutSmart buffer (10x) 5 µl 5 µl Alkaline phosphatase 1 µl -- DNA Up to 5 µg Up to 5 µg Nuclease-free H2O Add to 50 µl Add to 50 µl Total 50 µl 50 µl - Incubate at 37 °C for 1 h.
- During incubation, cast a 1% agarose gel to be used for isolating the digested products.
- Undertake gel electrophoresis with the entire volume of the restriction digest mixture.
Troubleshoot: it is advised to include a sample of the uncut and singly-cut IEX vector as controls. - Excise the doubly digested IEX vector (7.0 kb) and new CDS insert.
- Proceed to gel extraction purification per manufacturer’s protocol. Measure DNA concentration.
Day 1. Ligation of new CDS insert into doubly digested IEX vector- Determine the appropriate vector: insert mass ratios to achieve an approximate 1:1 molar ratio. This task can be simplified by using online algorithms, including the NEBioCalculator tool (version 1.10.0; https://nebiocalculator.neb.com/#!/ligation).
- Set up the following ligation reactions:
Reagents Vector-only control (µl) Vector + Insert (µl) T4 DNA ligation buffer (10x) 2 2 T4 DNA ligase 2 2 Vector (100 ng) Calculate Calculate Insert (x ng, depends on ratio) -- Calculate Nuclease-free H2O Add to 20 Add to 20 Total 20 20 - Incubate at room temperature for 15 min.
- Proceed to bacterial transformation per manufacturer’s protocol.
- Plate bacteria on agar plates with 100 µg/ml ampicillin (the same concentration is used for all applications involving ampicillin), incubate plates upside down at 30 °C overnight.
Endura chemically competent cells are used to minimize truncation of complex plasmids at 30 °C growth temperature on agar. Agar plates are incubated upside down to reduce evaporation of moisture from the agar during overnight incubations.
Day 2. Pick transformant colonies- Compare the number of colonies on the ‘vector-only’ control plate, which should have none to very few colonies, and the ‘vector plus insert’ plate, which should have many colonies.
- Pick 6-10 colonies from the ‘vector plus insert’ plate to grow in 3 ml of LB medium with ampicillin with shaking at 225 rpm overnight at 37 °C.
Day 3. Extract DNA for sequencing- Save 500 µl of the overnight bacteria culture as a glycerol stock.
- Extract the DNA from the remaining bacteria culture by the miniprep method as per manufacturer’s protocol.
- Measure the DNA concentration and send the purified DNA plasmid for sequencing.
- If additional site-directed mutagenesis is required, perform the reaction using the Q5® Site-directed mutagenesis kit according to manufacturer’s protocol.
Days 4-5. Maxiprep new IEX and iCre vectors for transfection- Use the bacteria stock with the correct sequencing result to grow in a 125-250 ml culture with ampicillin, then undertake a DNA maxiprep following manufacturer instructions.
Note: It is important to use a maxiprep kit that removes endotoxins from the bacteria to minimize undesired cellular toxicity during future transfections. - At the same time, maxiprep the iCre vector for future transfections.
- Week 2. Generating inducible cells with new IEX vector
Next steps are designed to create inducible cells by co-transfection of the IEX vector and the iCre vector, followed by puromycin selection and confirmation of inducibility by doxycycline (Dox) treatment.
Ahead of Day 8- Culture target cells in cell culture incubators at 37 °C, 95% moisture, and 5% CO2.
- Perform all protocols involving mammalian cells in certified biosafety cabinets to prevent contamination and adhere to biosafety rules and regulations.
- Passage at least once before transfection to check that cells are growing normally as they can be quite fragile after thawing. See ‘Auxiliary information–Cell culture’ section for instructions on regular cell culture, passaging, and saving stocks.
- Determine the appropriate puromycin concentration to use for selection for each cell line by treating cells at various concentrations (e.g., 0.0, 0.2, 0.5, 1.0, 2.0, 5.0, and 10 µg/ml) of puromycin and selecting the lowest concentration that kills 100% of cells in 3 days.
Day 8. Plate cells for transfection- Plate all cells in media without antibiotics or antimycotics.
- For ReN VM cells, plate ~135,000 cells/well on Matrigel-coated 12-well plates in proliferation media without heparin. Cells should be ~50% confluent the next day.
- For cells that do not need to be plated on coated cell culture dishes, plate ~350,000 cells/well on 6-well plates with the expectation that they are ~80% confluent the next day.
- Set 1-2 wells aside as controls for effective puromycin selection; they will not be transfected.
Day 9. Transfection- Transfect cells using a ratio of 1 iCre vector: 4 IEX vectors with desired insert.
A ratio of 1 iCre vector: 1-3 IEX vectors have also worked. - For ReN VM cells, use a total of 1.0 μg of DNA with 1 µl of TransfeX transfection reagent per well following manufacturer’s instructions.
Use proliferation media without heparin during the transfection because heparin may compromise effectiveness. - For most other cells, use a total of 3 µg of DNA with jetPrime transfection reagent per well following manufacturer’s instructions.
For IMR-32 cells, use a ratio of 1 µg DNA:3 µl jetPrime.
For HEK-293 cells, use a ratio of 1 µg DNA:2 µl jetPrime. - To minimize toxicity change media 4 h after transfection to regular proliferation media.
- Week 3. Puromycin selection and Dox treatment
- Passage transfected cells onto a few larger plates and add the appropriate amount of puromycin and 2.0 µg/ml of Dox.
One may check for induction levels 24 h post Dox addition on a fluorescence microscope. - Continue puromycin selection until all cells in the control wells have died.
Note: It is recommended to refresh puromycin and Dox every other day to keep the inducible transgene transcriptionally active. - Allow surviving cells to proliferate to sizable colonies of > 100 cells/colony.
- Induce cells with 2.0 µg/ml Dox; check on the fluorescence microscope for successfully induced colonies.
Caution: Induction in ReN VM cells may not be homogenous even within the same clone and may lose induction during the course of differentiation. This is because endogenous epigenetic silencing may cause variable and mosaic expression of inducible constructs (Chanda et al., 2017). Also, if differentiation is desired, induction of ReN VM cells and other neural progenitor cell lines may be improved by beginning induction 2-4 days during the proliferation stage before the start of differentiation. - Mark and transfer selected colonies with desired level of induction to new wells. Expand them to save as stocks and to validate the inducibility and identity of the protein-of-interest by Western blot.
- If removal of the PuroR gene is desired, transfect cells with the ‘Flpo’ optimized Flippase vector.
- Select cells that have lost puromycin resistance by plating duplicates of the same cell clone, one of them puromycin treated the other left untreated.
In ReN VM cells, retaining PuroR may help with keeping the inducible transgene region transcriptionally active.
- Passage transfected cells onto a few larger plates and add the appropriate amount of puromycin and 2.0 µg/ml of Dox.
- Week 4. Preparing inducible cells for mass spectrometry-based interactome studies
Note: The following steps are designed to capture EGFP-tagged inducible protein and its interactors using GFP nanotrap for downstream analysis in mass spectrometer.- Culture inducible protein-of-interest-EGFP fusion cells in biological triplicates on 15-cm plates along with cells inducibly expressing EGFP only as a control against unspecific binding to the GFP nanotrap matrix.
Note: If induced expression is very low, scale up accordingly. - Induce cells for the desired length of time.
- Follow manufacturer’s protocol on using GFP nanotrap for affinity capture experiments.
All procedures should be performed at 4 °C to prevent protein degradation. - Cells may be lysed in RIPA buffer as per manufacturer’s protocol or buffer composed of 0.5% NP-40 and 0.25% DOC (Recipe 4).
Remove cellular debris by centrifugation at 3,000 x g for 5 min at 4 °C. - Due to its high binding capacity, 20 μl of GFP nanotrap bead slurry/biological replicate should be sufficient for mass spectrometry-based interactome studies.
- EGFP fusion proteins or EGFP alone may be captured on GFP nanotrap beads during 2 h incubations at 4 °C with continuous mixing with the lysate.
Shorter nanotrap bead incubations of 5 min followed by light centrifugation may be sufficient to visualize the capture of EGFP fusion proteins. - Centrifuge at 100 x g at 4 °C to collect agarose-conjugated GFP nanotrap beads and bait proteins.
- Wash beads 5 times in Wash buffer. Washing procedures should be done within 2 h to minimize loss of interactors.
- First 3 times in Washing buffer (Recipe 5). NaCl salt concentration of 150-500 mM can be used during these washing steps, depending on desired stringency.
- The last two washes should be 25 mM HEPES (pH 7.0) and 10 mM HEPES (pH 7.0). Tris-HCl must not be used.
- Elute in 0.2% trifluoroacetic acid with 20% acetonitrile in water (pH 1.9). Immediately save and flash freeze or pH neutralize a portion of the eluates (e.g., 25% of total) for downstream validation of bait protein complexes by Western blot.
A delay in the collection of the eluate material dedicated to Western blot analysis can lead to precipitation of eluate proteins in the pH 1.9 environment, thereby compromising the Western blot analysis. - Other special considerations:
- One may use BCA to adjust for equal protein content before affinity capture.
- Remember to save a fraction of the cellular lysates as ‘input’ and ‘unbound’ for Western blot analyses.
Auxiliary information
Cell culture- After thawing cells at 37 °C, remove DMSO content from the cell stock by diluting the cell stock in 4 ml of proliferation media and centrifuging cells at 100 x g for 3 minutes at 25 °C. Remove the supernatant to retain only the cell pellet. DMSO may be toxic to cells in regular culture.
- Culture cells in the appropriate proliferation media for about one week to allow recovery from thawing.
- For ReN VM cells, do not include antibiotics or antimycotics in the proliferation media, as they will cause cell death.
- For ReN VM cells, please coat the tissue-culture plates with Matrigel according to the manufacturer’s protocol at least 2 h before seeding cells onto the plates. 2 h is needed for Matrigel to solidify at 37 °C in a humidified incubator.
- Passage cells regularly before they reach 90% confluency to prevent the adverse effects of overgrowing with the appropriate dissociation reagent. For ReN VM cells, use Accutase. For most other cell types, use trypsin.
- Remember to save a portion of the cells, especially valuable clones, as stocks and while using the rest for experiments or regular culture.
- For preserving cell stocks, resuspend cells in Cell Freezing Media in labelled cryovials. Transfer cells to -80 °C immediately and store at -80 °C for 1 day before transferring into liquid nitrogen for long term storage.
- Culture inducible protein-of-interest-EGFP fusion cells in biological triplicates on 15-cm plates along with cells inducibly expressing EGFP only as a control against unspecific binding to the GFP nanotrap matrix.
Data analysis
Information on statistical analyses as well as were to find publicly available mass spectrometry data from the TAU and BAG5 analyses was included with the article describing the first use of this system (Wang et al., 2019), with Figure 2 of the original article providing details on the design of a quantitative interactome analysis, Figure 3 describing mass spectrometry sequence coverage of the TAU protein, Figure 6 comparing the TAU interactome in IMR32 and ReN VM cells, and Figure 9 documenting results from a post-translational modification analysis of TAU produced with this system.
Recipes
- Proliferation media for IMR-32 and HEK-293 cells
Note: Make media fresh, store at 4 °C, stable for 2 months.
DMEM media with high glucose, L-glutamine, and sodium pyruvate supplemented with 10% qualified fetal bovine serum and 1% GlutaMAXTM-I supplement - Proliferation media for ReN VM cells
Note: Make media fresh, store at 4 °C, stable for 2 weeks.
DMEM/F12, 1:1 with L-glutamine but no HEPES supplemented with 2% N21-MAX supplement or 2% B-27 serum-free supplement
20 ng/ml basic fibroblast growth factor
20 ng/ml epidermal growth factor
2 ng/ml heparin - Neuronal differentiation media for ReN VM cells
Note: Make media fresh, store at 4 °C, stable for 1 month.
DMEM/F12 1:1 with L-glutamine but no HEPES supplemented with 2% N21-MAX supplement or 2% B-27 serum-free supplement
2 ng/ml glial-derived neurotrophic factor
500 μM dibutyryl-cyclic-adenosine monophosphate - Lysis buffer
Note: Make fresh before use, stable at 4 °C for 1 day.- Mix 0.5% (v/v) NP-40, 0.5% (w/v) DOC, Tris/HCl, pH 8.3, 5 mM EDTA, 1 mM Na3VO4, 10 mM NaF, 1 mM PMSF, 1x cOmplete protease inhibitor, and 1x PhosStop phosphatase inhibitor
- Prepare NP-40 and DOC as 50x stock solutions in water and warm gently to dissolve. DOC will precipitate at pH value below 8.0. Therefore, ensure that the pH exceeds this value and is buffered before adding this detergent
- Wash buffer
Note: Make fresh before use, stable at 4 °C for 2 days.
The composition of this buffer is similar to the lysis buffer with the following considerations:
Protease and phosphatase inhibitors can be omitted.
If samples are used for mass spectrometry downstream, the last two washes must use only 25 mM HEPES (pH 7.0) and 10 mM HEPES (pH 7.0) in deionized water. Tris-HCl must not be used. - Elution buffer
Note: Make fresh before use, stable at room temperature for 1 day.- Mix 0.2% TFA and 20% acetonitrile in deionized water
- The pH of this solution will be 1.9 without adjustment
Acknowledgments
XW held an Ontario Graduate Scholarship. GS received funds from the Krembil Foundation, Canadian Institutes for Health Research (grant number 137651) and the Alberta Prion Research Institute (grant number 201600028). Support by the Arnold Irwin family for the purchase of a mass spectrometer was instrumental for getting started on this project. The authors gratefully acknowledge generous philanthropic support by the Borden Rosiak family.
Competing interests
The authors declare that they have no financial or non-financial competing interests.
Ethics
The study did not involve human subjects or animal work. All handling of human cell lines and chemicals were undertaken in accordance with an active biosafety protocol (number 208-S06-2) approved by the environmental health and safety (EHS) office at the University of Toronto, Toronto, Canada.
References
- Chanda, D., Hensel, J. A., Higgs, J. T., Grover, R., Kaza, N. and Ponnazhagan, S. (2017). Effects of cellular methylation on transgene expression and site-specific integration of adeno-associated virus. Genes (Basel) 8(9): 232.
- DeKelver, R. C., Choi, V. M., Moehle, E. A., Paschon, D. E., Hockemeyer, D., Meijsing, S. H., Sancak, Y., Cui, X., Steine, E. J., Miller, J. C., Tam, P., Bartsevich, V. V., Meng, X., Rupniewski, I., Gopalan, S. M., Sun, H. C., Pitz, K. J., Rock, J. M., Zhang, L., Davis, G. D., Rebar, E. J., Cheeseman, I. M., Yamamoto, K. R., Sabatini, D. M., Jaenisch, R., Gregory, P. D. and Urnov, F. D. (2010). Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res 20(8): 1133-1142.
- Devkota, S. (2018). The road less traveled: strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Cas-mediated transgenesis. BMB Rep 51(9): 437-443.
- Donato, R., Miljan, E. A., Hines, S. J., Aouabdi, S., Pollock, K., Patel, S., Edwards, F. A. and Sinden, J. D. (2007). Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci 8: 36.
- Ran, F. A., Hsu, P. D., Lin, C. Y., Gootenberg, J. S., Konermann, S., Trevino, A. E., Scott, D. A., Inoue, A., Matoba, S., Zhang, Y. and Zhang, F. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6): 1380-1389.
- Rothbauer, U., Zolghadr, K., Muyldermans, S., Schepers, A., Cardoso, M. C. and Leonhardt, H. (2008). A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 7(2): 282-289.
- Wang, X., Williams, D., Müller, I., Lemieux, M., Dukart, R., Maia, I. B. L., Wang, H., Woerman, A. L. and Schmitt-Ulms, G. (2019). Tau interactome analyses in CRISPR-Cas9 engineered neuronal cells reveal ATPase-dependent binding of wild-type but not P301L Tau to non-muscle myosins. Sci Rep 9(1): 16238.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wang, X., Friesen, E., Müller, I., Lemieux, M., Dukart, R., Maia, I. B., Kalia, S. and Schmitt-Ulms, G. (2020). Rapid Generation of Human Neuronal Cell Models Enabling Inducible Expression of Proteins-of-interest for Functional Studies. Bio-protocol 10(9): e3615. DOI: 10.21769/BioProtoc.3615.
Category
Neuroscience > Development > Neuron
Biochemistry > Protein > Fluorescence
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.