- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Enterovirus Competition Assay to Assess Replication Fitness

Published: Vol 9, Iss 10, May 20, 2019 DOI: 10.21769/BioProtoc.3233 Views: 6119
Reviewed by: Kristin L. ShinglerJean Kaoru Millet
Original research article
The authors used this protocol in:

Nov 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
In virology the difference between the fitness of two viruses can be determined by using various methods, such as virus titer, growth curve analysis, measurement of virus infectivity, analysis of produced RNA copies and viral protein production. However, for closely performing viruses, it is often very hard to distinguish the differences. In vitro competition assays are a sensitive tool for determining viral replication fitness for many viruses replicating in cell culture. Relative viral replication fitness is usually measured from multiple cycle growth competition assays. Competition assays provide a sensitive measurement of viral fitness since the viruses are competing for cellular targets under identical growth conditions. This protocol describes a competition assay for enteroviruses and contains two alternative formats for initial infections, which can be varied depending on specific goals for each particular experiment. The protocol involves infection of cells with competing viruses, passaging, RNA extraction from infected cells, RT-PCR and Sanger sequencing followed by comparative analysis of resulting chromatograms obtained under various initial infection conditions. The techniques are applicable to members of many virus families, such as alphaviruses, flaviviruses, pestiviruses, and other RNA viruses with an established reverse genetics system.
Keywords: EnterovirusBackground
Enteroviruses comprise a large group of mammalian pathogens that includes poliovirus. Pathology in humans ranges from sub-clinical to acute flaccid paralysis, myocarditis and meningitis. In our recent paper (Lulla et al., 2019) we reported that many enterovirus genomes harbor an upstream open reading frame (uORF) that encodes an additional viral protein, UP (upstream protein). Using the echovirus 7 (EV7) reverse genetics system, we created two UP knock-out mutants (EV7-Loop and EV7-PTC), which exhibited wtEV7-like growth properties. Therefore, we decided to perform a competition assay to determine possible subtle fitness defects.
Materials and Reagents
- Materials
- Pipette tips (Fisher, catalog numbers: 02-707-426, 02-707-403, 02-707-438)
- 1.5 ml microfuge tubes (STARLAB, catalog number: 51615-5500)
- T175 tissue culture flasks (TPP® tissue culture flasks, Sigma, catalog number: Z707562)
- 12-well polystyrene culture plates (TPP® tissue culture plates, Sigma, catalog number: Z707775)
- Viruses
Echovirus 7 (EV7), derived from EV7 infectious clone
Note: The cDNA of Echovirus 7 strain Wallace was sourced from Michael Lindberg (GenBank accession number AF465516, with the silent substitution 1687G-to-A) and was cloned downstream of a T7 RNA promoter. Mutant viruses EV7-Loop and EV7-PTC were generated by mutagenesis of the original EV7 infectious clone and prepared exactly as wt EV7 (Lulla et al., 2019). - Cell lines
RD cells (human rhabdomyosarcoma cell line, ATCC, CCL-136), verified mycoplasma-free by next-generation sequencing - Reagents
- Dulbecco’s modified Eagle’s medium (DMEM) (Sigma, catalog number: D6546)
- Fetal bovine serum (FBS) (PAN Biotech, catalog number: P40-37500)
- Penicillin-streptomycin (10,000 U/ml) (Life Technologies, catalog number: 15140-122)
- 200 mM L-Glutamine (Gibco, catalog number: 25030081)
- Phosphate buffered saline (PBS) (Life Technologies, catalog number: 14190-144)
- 1 M HEPES buffer, pH range: 7.2-7.5 (Life Technologies, catalog number: 15630-080)
- 0.25% Trypsin (Gibco, catalog number: 15090046-100)
- Bovine serum albumin (BSA) (PAN Biotech, catalog number: P06-1395500)
- Direct-zolTM RNA MiniPrep Plus (Zymo Research, catalog number: R2072)
- PhusionTM RT-PCR Kit (Thermo Scientific, catalog number: F-546S)
- Complete DMEM (see Recipes)
- Serum-free DMEM (see Recipes)
Equipment
- -80 °C freezer
- Vortexer
- Pipettes: 1000 µl, 200 µl, 20 µl, 2 µl
- CO2 incubator
- Haemocytometer
- BSL2 cell culture cabinet
- Rocking platform shaker
- Phase-contrast inverted microscope (Nikon TMS)
- 4 °C refrigerator
- Autoclave
Software
- BioEdit (Free software, Ibis Therapeutics, http://www.mbio.ncsu.edu/BioEdit/bioedit.html)
Procedure
IMPORTANT NOTE: All experiments with viruses (i.e., all pre-Trizol RNA isolation steps) should be performed inside a biosafety level 2 (BSL2) tissue culture laboratory according to the country and institution regulations and required permits regarding enterovirus strains handling and storage.
- Infection and virus passaging
- Prepare cells for infection 1 day before by seeding 3 x 105 RD cells per well on 12-well plates (the aim is to achieve 5 x 105 RD cells per well by the next day, which corresponds to subconfluent RD monolayers).
- RD cells seeded on T175 flasks and grown in complete DMEM until 80-90% confluency. Aspirate media from the cells and rinse with 10 ml of PBS. Trypsinize RD cells using 3 ml of 0.25% trypsin by gently tapping the flask.
- Once cells are detached, add 10 ml of complete DMEM, ensure the cells are properly resuspended by pipetting up and down. Count cells in suspension using hemocytometer.
- Dilute cell suspension with complete DMEM to get concentration corresponding to 3 x 105 cells per 1 ml. Seed 1 ml of cell suspension to each well of 12-well plates.
- Incubate overnight in 37 °C, 5% CO2 incubator.
- Check the cell monolayers using a microscope (equal healthy cell monolayers, no CPE). Remove media from cell monolayers and infect with a total multiplicity of infection (MOI) of 0.1, in 0.2 ml of serum-free DMEM. Use the following formula to calculate the amount of virus needed:

Example: For 109 PFU/ml virus stock use, 5 x 105 cells x 0.1 PFU/cell x 103 μl/ml/109 PFU/ml = 0.05 μl. - Mix using wt EV7 and mutant at either equal or 1:9 proportions, with a total MOI 0.1.
Note: This assay can be completed with other viruses and cell lines. Different proportions and MOI can be used for this experiment depending on the virus growth kinetics and susceptibility of cells for infection. Using MOI 0.1 means that more than 90% of the cells remain uninfected providing enough space for competing viruses; any growth difference should also accumulate over passaging. - Use triplicates for each infection, monoinfections for positive controls and mock (uninfected) cells as negative controls as suggested in Figure 1.
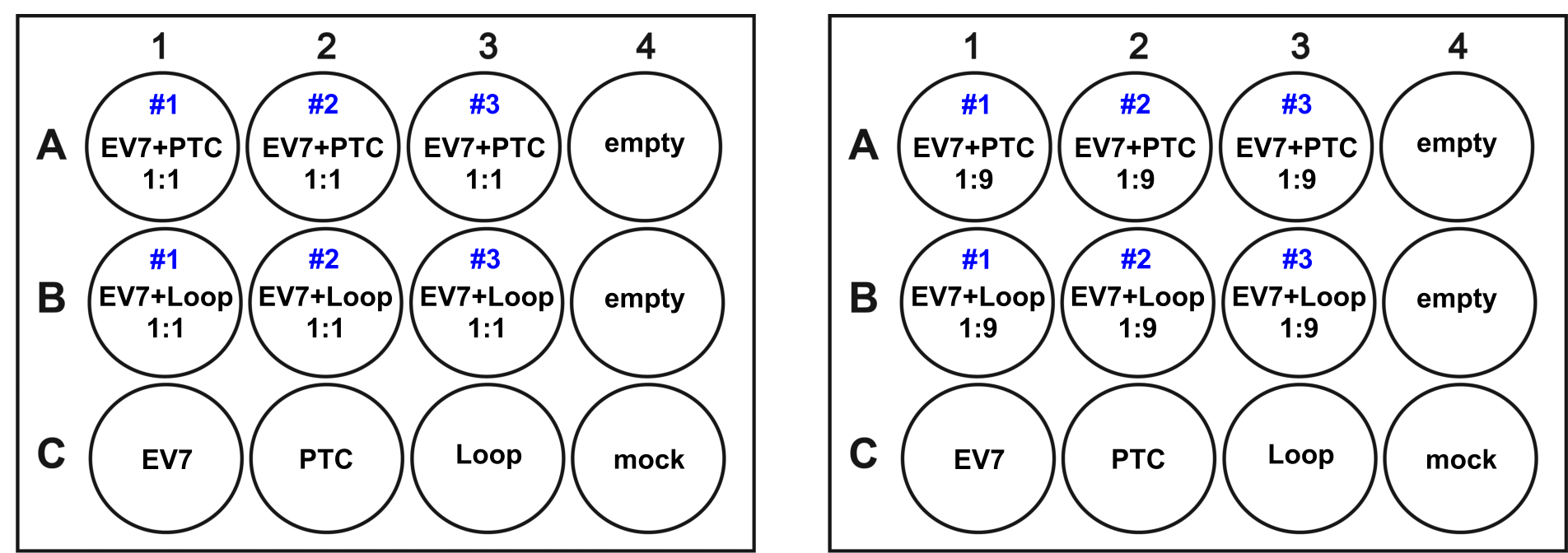
Figure 1. Sample layout of two 12-well plates at either equal (Left) or 1:9 (Right) proportions. Every subsequent passage is performed on similarly aranged 12-well plates. - Incubate infected cells at room temperature on a gently rocking platform for 1 h. To remove unbound virus, aspirate infection media, wash each well with 1 ml of serum-free DMEM, and overlay cells with 0.5 ml of serum-free DMEM.
- Incubate at 37 °C in a CO2 incubator overnight (16-20 h) until complete CPE is observed.
- Collect media from infected plates to a 1.5 ml tube, centrifuge at 9,600 x g for 5 min, then transfer to the new 1.5 ml tube and measure the volume.
- Add 0.2% BSA to supernatant (10 μl of 10% BSA per 0.5 ml of clarified supernatant).
- Perform 5 blind passages (with estimated, but unknown titer) using 1:10,000 volume of obtained virus stock (corresponds to MOI 0.05-0.2). Use the same infection protocol as before (steps 1-8).
Note: For less efficiently replicating viruses more than 5 passages could be needed. - Store virus stocks at -80 °C.
- Add 1 ml PBS to remaining cells and collect them by pipetting into a 1.5 ml tube (infected cells detach easily and do not require scraping). Centrifuge cells at 9,600 x g for 5 min, remove supernatant and proceed to RNA isolation from the infected cells.
- Prepare cells for infection 1 day before by seeding 3 x 105 RD cells per well on 12-well plates (the aim is to achieve 5 x 105 RD cells per well by the next day, which corresponds to subconfluent RD monolayers).
- RNA isolation from infected cells
- Isolate RNA from passages 1 and 5 using Direct-zolTM RNA MiniPrep Plus (Zymo Research) according to the manufacturer’s protocol (Direct-zolTM RNA MiniPrep Plus INSTRUCTION MANUAL ver.1.10.1).
- Perform RT-PCR using Thermo ScientificTM PhusionTM RT-PCR Kit using virus-specific primers and manufacturer’s recommendations (Thermo Scientific Phusion RT-PCR Kit manual). The example design of the introduced mutations and sequencing primer is presented in Figure 2.
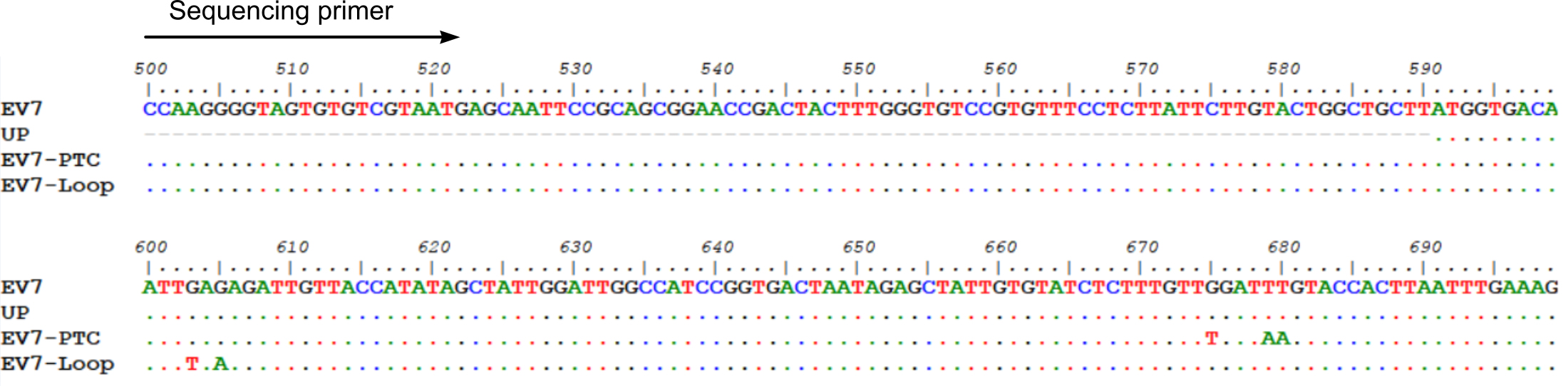
Figure 2. Example design of mutant virus genomes and sequencing primer. Depending on sequencing facilities, a 300-1000 bp long PCR fragment should be optimal for this assay. - Perform Sanger sequencing of the PCR fragment containing the mutated region of the virus genome.
Note: Please consider the nature of the virus (intracellular or budded virions) for choosing the correct sample to analyze. Sometimes analyzing RNA from media rather than from infected cells can be considered. For enteroviruses it is not a crucial step; therefore an easier approach (RNA isolation from infected cells) was chosen.
Data analysis
Compare chromatograms and evaluate based on three RT-PCR products from each analyzed sample (Figure 3). In the experimental conditions tested viruses do not differ in their growth properties. The chromatograms should shift if the viruses differ in their growth properties. Compare the peak areas from at least three different chromatograms for each sample and estimate the changes. If wt virus out-competes the mutant, the chromatogram of the fifth passage sample should match the wt virus chromatogram.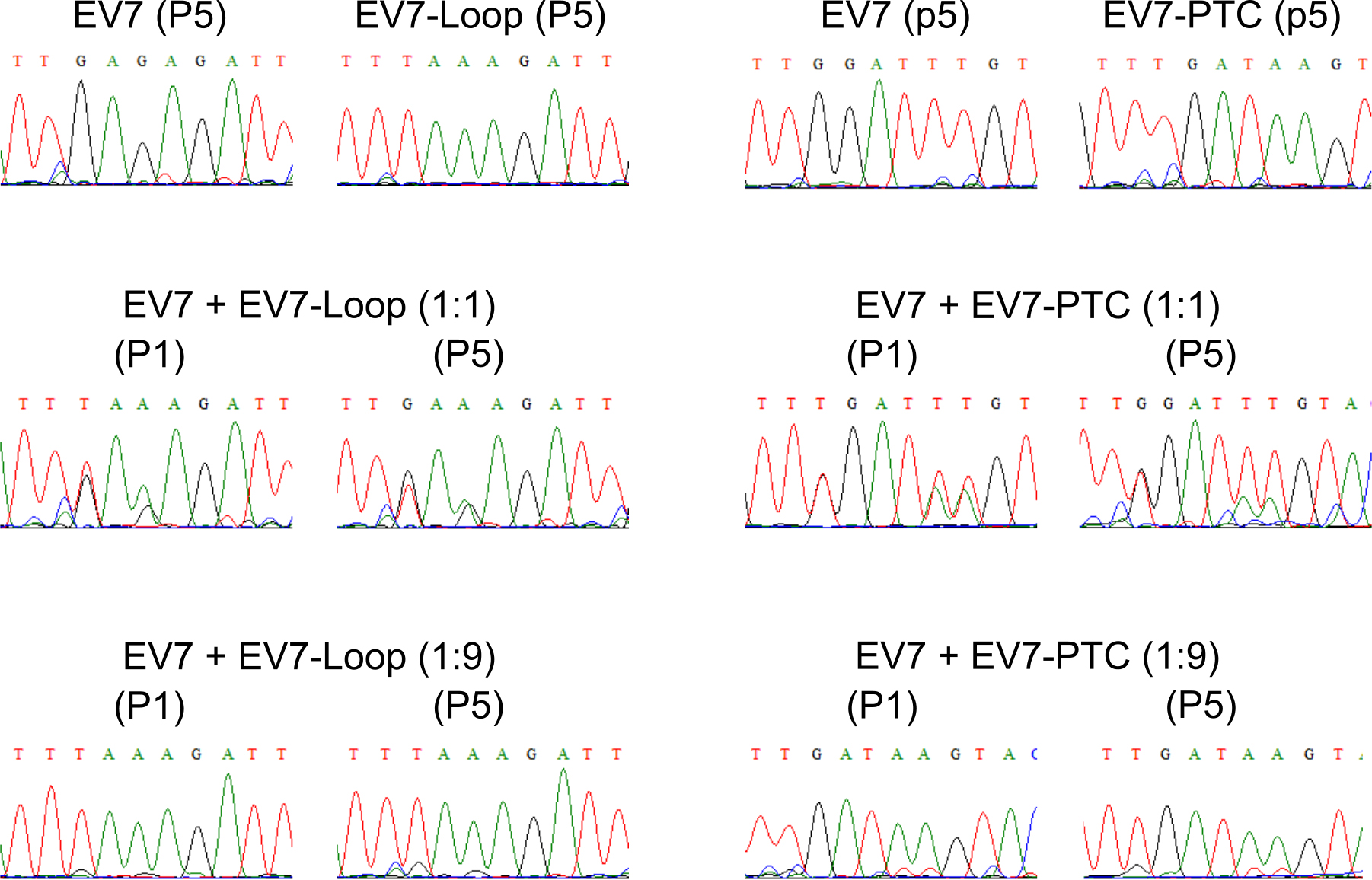
Figure 3. Sample sequencing chromatograms of RT-PCR products for the competition. Experiment of EV7, EV7-PTC and EV7-Loop mutants at initial infection (P1) and after five passages (P5). The figure is reproduced from Supplementary Figure 3 in Lulla et al. (2019).
Recipes
- Complete DMEM
500 ml DMEM
50 ml FBS
5 ml 200 mM L-Glutamine
5 ml penicillin-streptomycin (10,000 U/ml)
10 ml 1 M HEPES pH 7.2-7.5
Store at 4 °C - Serum-free DMEM
500 ml DMEM
5 ml 200 mM L-Glutamine
5 ml penicillin-streptomycin (10,000 U/ml)
10 ml 1 M HEPES, pH 7.2-7.5
Store at 4 °C
Acknowledgments
This work was supported by Wellcome Trust grant [106207] and European Research Council grant [646891] to A.E.F. The cDNA of Echovirus 7 strain Wallace was sourced from Michael Lindberg. We thank Hazel Stewart for critical reading of the protocol.
Competing interests
The authors declare that they do not have any conflicts of interests or competing interests.
References
- Lulla, V., Dinan, A. M., Hosmillo, M., Chaudhry, Y., Sherry, L., Irigoyen, N., Nayak, K. M., Stonehouse, N. J., Zilbauer, M., Goodfellow, I. and Firth, A. E. (2019). An upstream protein-coding region in enteroviruses modulates virus infection in gut epithelial cells. Nat Microbiol 4(2): 280-292.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lulla, V. and Firth, A. E. (2019). Enterovirus Competition Assay to Assess Replication Fitness. Bio-protocol 9(10): e3233. DOI: 10.21769/BioProtoc.3233.
Category
Microbiology > Microbial cell biology > Virus propagation
Molecular Biology > RNA > RNA purification
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.