- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantification of Cutaneous Ionocytes in Small Aquatic Organisms

Published: Vol 9, Iss 9, May 5, 2019 DOI: 10.21769/BioProtoc.3227 Views: 5891
Reviewed by: Gal HaimovichMichelle GoodyYong Teng
Original research article
The authors used this protocol in:

Feb 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Aquatic organisms have specialized cells called ionocytes that regulate the ionic composition, osmolarity, and acid/base status of internal fluids. In small aquatic organisms such as fishes in their early life stages, ionocytes are typically found on the cutaneous surface and their abundance can change to help cope with various metabolic and environmental factors. Ionocytes profusely express ATPase enzymes, most notably Na+/K+ ATPase, which can be identified by immunohistochemistry. However, quantification of cutaneous ionocytes is not trivial due to the limited camera’s focal plane and the microscope’s field-of-view. This protocol describes a technique to consistently and reliably identify, image, and measure the relative surface area covered by cutaneous ionocytes through software-mediated focus-stacking and photo-stitching–thereby allowing the quantification of cutaneous ionocyte area as a proxy for ion transporting capacity across the skin. Because ionocytes are essential for regulating ionic composition, osmolarity, and acid/base status of internal fluids, this technique is useful for studying physiological mechanisms used by fish larvae and other small aquatic organisms during development and in response to environmental stress.
Keywords: Chloride cellBackground
Ionocytes (formerly called chloride cells and mitochondrion-rich cells) are specialized cells that transport ions to regulate the ionic composition, osmolarity, and acid/base status of internal fluids (reviewed in Evans et al., 2005). Ionocytes typically express high levels of the enzyme Na+/K+ ATPase, which can be used to visualize them using immunohistochemical techniques. In larval and juvenile fishes, ionocytes are present throughout the skin (reviewed in Varsamos et al., 2005). The abundance, distribution, and size of ionocytes can change during development, as well as in response to environmental stressors such as temperature, acidification, salinity, hypoxia, UV light, and pollutants. However, accurate and consistent quantification of cutaneous ionocytes can be difficult due to their presence in large numbers (tens of thousands), non-homogenous distribution, and being masked by pigmented cells. In addition, ionocyte quantification throughout the entire skin of an organism is often limited by the microscope’s field-of-view (Fridman et al., 2011) and both the camera’s field-of-view and focal plane (Hiroi et al., 1999; Varsamos et al., 2002). Because ionocyte distribution is often quite variable across the larval skin, estimations of ionocyte abundance based on a single photograph and focal plane may not be representative of the organism as a whole, and therefore not reliable or repeatable. As such, previous studies that utilized immunohistochemistry to identify and quantify ionocytes have been descriptive in nature (Hwang, 1989; Hiroi et al., 1998; Katoh et al., 2000), which precludes replication and comparison across species and treatments.
Here, we describe a protocol involving immunohistochemistry, photo-microscopy, and photo-editing software that allows estimating ionocyte number, size, density, and relative coverage of cutaneous ionocytes with high accuracy. Unlike previous studies, our method resolves both frame-of-view and focal plane limitations through focus-stacking and photo-stitching software, effectively digitizing a 3-D fish larva into a 2-D image. Additionally, the resulting digital image retains its high-resolution–thereby allowing the accurate quantification of all ionocytes on one side of the larva along with their average size and the larva’s surface area. These parameters can then be inputted into a simple equation to estimate the cutaneous ionocyte ion-transporting capacity in relation to the total skin surface area of the specimen. For larger specimens in which counting all cutaneous ionocytes is cumbersome and time-consuming, we developed a random sampling protocol that allows highly accurate cutaneous ionocyte estimations after counting ionocytes within 10% of the skin surface area. For example, the estimated number of cutaneous ionocytes in a 13.7 mm long Yellowfin Tuna (Thunnus albacares) specimen was 15,342, which was only 3.6% higher than the actual 14,795 cutaneous ionocytes counted in the entire fish skin (Kwan et al., 2019). We believe this method will be valuable for future studies quantifying cutaneous ionocytes in larval and juvenile fish as well as other small aquatic organisms during development and in response to various environmental stress (e.g., ocean warming, hypoxia, and acidification).
Materials and Reagents
- PCR Reaction Strips (8 x 0.2 ml) attached flat cap (Simport, catalog number: T320-2N)
- 1.7 ml microcentrifuge tubes (Corning, catalog number: MCT-175-C)
- 15 ml plastic tubes (Corning Brand, catalog number: 352196)
- Microscope slides (Thermo Fisher Scientific, catalog number: 12-550-15) or concave microscope slides (United Scientific Supplies, catalog number: CS3X11)
- Microscope coverslips (e.g., VWR, catalog number: 48366 045)
- Microscope small Petri dish trays (MatTek Corp, catalog number: P35GC-1.5-10-C)
- Petri dish (Corning Brand, catalog number: 351029) or any other container
- Specimen of interest
Note: This protocol is optimized for small (< 30 mm) samples with relatively low natural pigmentation and. high transparency. This protocol could be used for other specimens, but additional optimization of immunostaining and image acquisition might be required. - Deionized (DI) water
- 10x Phosphate buffer saline (PBS) (Corning Incorporated, catalog number: 46-013-CM)
- 20% paraformaldehyde (PFA) (Electron Microscopy Sciences, catalog number: 15713)
- 200-proof (100%) Ethyl alcohol (Decon Laboratories, catalog number: 2701)
- 30% (w/v) hydrogen peroxide (Sigma-Aldrich, catalog number: H3410)
- α5 Na+/K+ ATPase (NKA) antibody (Developmental Studies Hybridoma Bank, catalog number: a5–supernatant)
Notes:- This monoclonal antibody has specifically recognized NKA from multiple classes of aquatic organisms including: bony fishes (Wilson et al., 2000 and 2002; Yang et al., 2013; Tang et al., 2014; Kwan et al., 2019), elasmobranch fishes (Piermarini and Evans, 2001; Roa et al., 2014; Roa and Tresguerres, 2017), and hagfish (Clifford et al., 2015).
- This protocol may be adapted for other proteins following validation of primary antibodies in the species of interest. However, successful localization using different primary antibodies may require optimizing the fixation and immunohistochemical procedures described here.
- Vectastain® Elite® ABC HRP ready-to-use kit (Vector Laboratories, catalog number: PK-7200), includes:
- Normal horse serum blocking solution
- Pan-specific secondary antibody solution
- Streptavidin peroxidase solution
- 3,3-diaminobenzidine (DAB) Peroxidase Substrate stored at 4 °C (Vector Laboratories, catalog number: SK-4100)
- Sodium azide (Thermo Fisher Scientific, catalog number: S227I-25)
- 1.2x phosphate buffer saline (PBS) solution (see Recipes)
- 4% paraformaldehyde (PFA) fixative solution (see Recipes)
- 3% hydrogen peroxide solution (see Recipes)
- 1.0x PBS solution (see Recipes)
- DAB substrate working solution (see Recipes)
- 10% sodium azide stock solution (see Recipes)
- 0.02% sodium azide working solution (see Recipes)
Equipment
- Fridge (4 °C) and freezer (-80 °C)
- Chemical fume hood
- Borosilicate glass vial (Thermo Fisher Scientific, catalog number: 033374)
- Shake table (e.g., VWR, Standard Orbital Shaker, Model 1000) or rotator (e.g., Thermo Fisher Scientific, Tube Revolver/Rotator, catalog number: 88881001)
- Transfer pipette (Thermo Fisher Scientific, catalog number: 1368050) or any other pipette suitable for your specimen of interest
- Fine forceps (Fine Science Tools, catalog number: 26029-10) or any forceps that does not damage your specimen of interest
- Camera-mounted microscope(s) Leica DMR compound microscope for smaller samples (< 5 mm SL), Leica MZ9.5 stereomicroscope for larger samples (> 5 mm)
- Microscope light source (Ludl Electronic Products [LEP] [12 V, 100 W], catalog number: 99019)
- Microscope mounted camera (Canon, Rebel T3i single lens reflex camera)
- Compatible camera with SD card
- Anti-vibration table for microscopy imaging
- Microscope stage micrometer standard
- Monitor for microscope camera’s live-feed imaging
- Camera remote for wireless shutter release (Canon RC-6)
Software
- Helicon Focus (HeliconSoft, Version 6.7.2 or later, www.heliconsoft.com)
- Adobe Photoshop (Adobe Systems, version CS3 or higher, www.adobe.com)
- Fiji (https://imagej.net/Fiji), or ImageJ (https://imagej.nih.gov/ij/download.html) with cell counter plugin (https://imagej.nih.gov/ij/plugins/cell-counter.html)
- Adobe Illustrator (Adobe Systems, Version 19.2.0 or later, www.adobe.com)
- R (https://www.r-project.org)
Procedure
An overview of this protocol is shown below (Figure 1). The time necessary to finish this procedure varies with the size of each sample. At the minimum, this procedure will take at least 3 days.
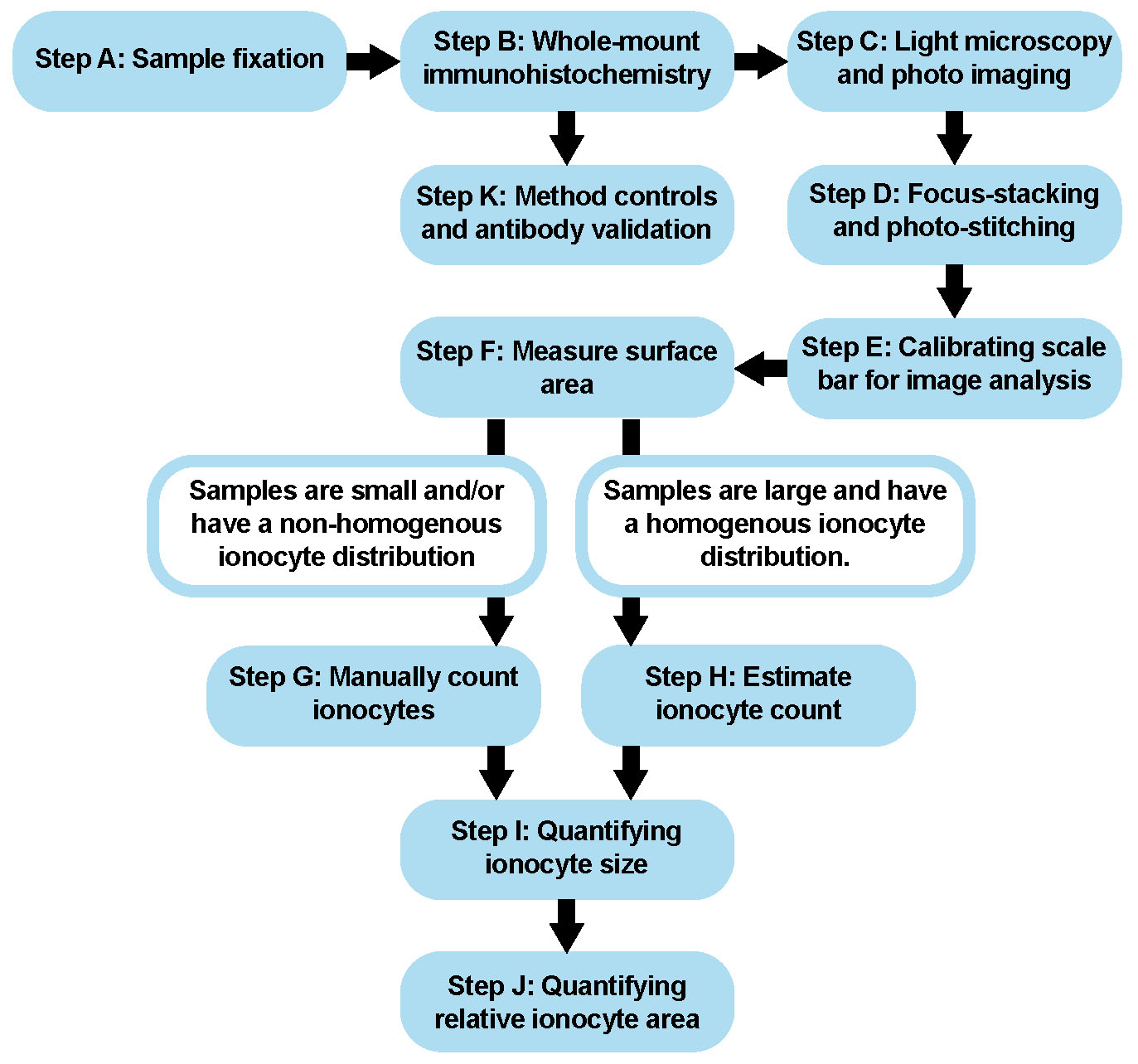
Figure 1. Procedural Overview. Schematic showing the major steps of this protocol.
- Sample fixation
- Euthanize specimen in accordance with an approved animal care protocol.
- Incubate samples in fixative solution (see Recipes 1 and 2) in any water-tight container (e.g., borosilicate glass vial) on a rotator/shake table overnight (8-12 h) at 4 °C.
Notes:- All samples should be fully immersed in the fixative. Multiple samples can be fixed together. In Kwan et al., 2019, 6-10 fish larvae (ranging from 3-25 mm in length) were fixed in 10 ml of fixative within a 20 ml borosilicate glass vial. Adjust container, volume, and fixative according to your specimen of interest. Rinsing the samples prior to fixation is not required.
- The ethanol washes (Steps A3 and A4) can be replaced with three 10 min PBS washes if the samples are to be immediately processed for immunohistochemistry.
- Remove fixative, then incubate samples in 50% ethanol on a shake table overnight (8-12 h) at 4 °C.
- Remove 50% ethanol, then incubate samples in 70% ethanol for long-term storage at 4 °C.
- Whole-mount immunohistochemistry
- Begin rehydrating samples by transferring onto a Petri dish with tap water for 1 min.
- After initial rinse, transfer samples into a plastic tube (e.g., 0.2 ml PCR tube or 1.7 ml microcentrifuge tube depending on larval size) with tap water (filled to max volume) and further incubate for 10 min at room temperature on a rotator/shake table.
Notes:- All samples should be fully immersed in solution. In Kwan et al., 2019, fish larva (< 20 mm in length) were incubated in a 1.7 ml microcentrifuge tube and immersed in ~1 ml of solution. Larger samples will require a larger container and more solution (and vice versa). Adjust container and solution volume accordingly.
- During solution changes, it is preferable to remove the solution from the container–as opposed to moving the sample to containers with different solutions. This minimizes damage to the sample. This applies to all solution changes.
- If changing solution with a plastic transfer pipette becomes problematic, consider using a more appropriately sized pipette (P200 pipette with a 200 μl tip for 0.2 ml PCR tube and P1000 pipette with a 1,000 μl tip) for changing solutions. Adjust according to your sample and/or container.
- Remove water, then incubate samples in freshly prepared 3% hydrogen peroxide (see Recipe 3) for 10 min at room temperature on a rotator/shake table.
Notes:- During solution changes, it is preferable to remove the solution from the container–as opposed to moving the sample to containers with different solutions. This minimizes damage to the sample. This applies to all solution changes.
- This step may be extended to 1 h if there is intense pigmentation and/or non-specific background staining.
- Remove 3% hydrogen peroxide solution, then incubate samples in normal horse serum (NHS) blocking solution for 15 min at room temperature on a rotator/shake table.
- Prepare primary antibody solution (e.g., α5 NKA antibody: 20 ng/ml in NHS blocking solution).
- Remove NHS, then incubate samples in primary antibody solution and leave on a rotator/shake table overnight (≥ 8 h) at room temperature or (preferably) at 4 °C.
Note: This step may be modified for various antibody validation controls. See Procedure K for details. - Remove primary antibody solution, then rinse samples in 1x PBS (see Recipe 4) for 5 min at room temperature on a rotator/shake table. Repeat two more times for a total of three rinses.
- Remove 1x PBS wash, then incubate samples in pan-specific secondary antibody solution for 30 min at room temperature on a rotator/shake table.
- Remove pan-specific secondary antibody solution, then rinse samples in 1x PBS for 5 min at room temperature on a rotator/shake table. Repeat two more times for a total of three rinses.
- Remove 1x PBS wash, then incubate samples in streptavidin peroxidase solution for 15 min at room temperature on a rotator/shake table.
- Remove streptavidin peroxidase solution, then rinse samples in 1x PBS for 5 min at room temperature on a rotator/shake table. Repeat two more times for a total of three rinses.
- Prepare the DAB substrate working solution (see Recipe 5) according to the manufacturer’s instruction in a separate container (e.g., 15 ml centrifuge tube).
- Remove 1x PBS wash, then incubate samples in DAB substrate working solution for 20 s at room temperature.
Notes:- The sensitivity of this portion is highly dependent on the tissue sample and antibody of interest. Because DAB staining is irreversible, start with 20 s and increase incubation time if darker staining is necessary. Optimal incubation time should be determined by the investigator(s).
- Incubating samples for a longer duration may result in darker background staining. If this becomes an issue, consider diluting the DAB substrate working solution with DI water.
- Remove DAB substrate working solution, then rinse samples in DI water for 10 min at room temperature on a rotator/shake table. Repeat two more times for a total of three rinses.
- Store in DI water at 4 °C until imaging.
- For long-term storage (1 week or longer), store in PBS + 0.02% sodium azide (see Recipes 6 and 7).
Note: Sodium azide is very toxic. Samples incubated within sodium azide should be given multiple DI water washes before handling and imaging.
- Light microscopy and imaging
- Carefully transfer sample onto a microscope slide, concave microscope slide, or a glass bottom dish for examination under a light microscope.
- Add DI water to keep sample from drying out, and secure sample with a coverslip to prevent unintentional movement.
- Switch on the live feed option on the DSLR camera.
- Switch off the autofocus option on the DSLR camera.
- Set camera to manual or aperture priority mode. Adjust the following parameters until image is optimal: camera ISO, camera aperture, microscope aperture, light source brightness.
- For optimal imaging, use a wireless remote controller to trigger shutter release.
Note: Avoid bumping into the entire camera apparatus. Because the exposure time of each photograph will likely require ~1 s per image, any movement of the microscope, sample, camera, and surrounding can cause blurriness. - Starting at one end (e.g., head of fish specimen), photograph sample at each focal plane (Z-stack) within the field of view. Manually adjust the focus until the entire Z-stack is finished (Figures 2A-2E). Be careful to not move the image in the X or Y-dimension.
Notes:- The image may differ between the microscope lens and camera (shown on the live feed monitor). Always use the camera’s focal plane and image accordingly.
- Shooting the Z-stack in consecutive order (front to back or vice versa) is preferable as it may help during post-processing.
- Ensure that your sample does not drift in the x or y plane between images for proper post-processing.
- An exact Z-step (distance between each image) ranges greatly amongst samples. After each image, turn the fine focus until the next plane is in focus. In Kwan et al., 2019, most images took around 6-8 images per Z-stack.
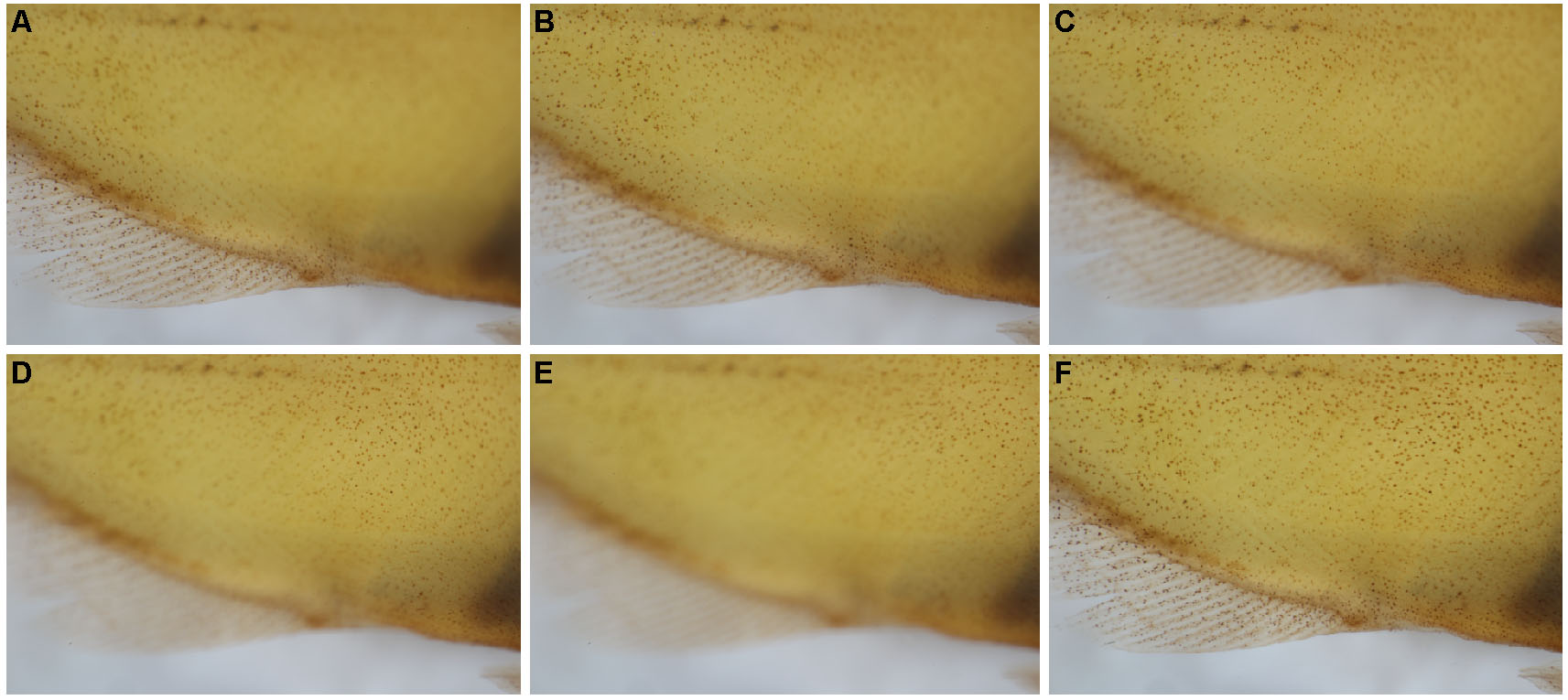
Figure 2. Example of Z-stacks and focus stacking. After photographing a Z-stack on the ventral body (A-E), the focused portions of each image were stacked together via focus stacking (F). - Move to the next unphotographed area, taking care to overlap consecutive Z-stacks by at least ~20% to ensure photo stitching accuracy (Figure 3).

Figure 3. Overlap of Z-stacks are necessary for proper photo-stitching. Each Z-stack (A, B) should have ~20% of overlapping area for optimal photo-stitching (C). - Continue imaging Z-stacks until the entire sample has been photographed.
- Photograph the microscope standard at a height equal to approximately half of the sample’s thickness.
Note: The same scale bar can be applied across multiple samples as long as the magnification of the microscope has not changed and the sample thickness is relatively similar.
- Focus-stacking and photo-stitching
Notes:- A video tutorial explaining the focus-stacking process can be found at youtube–Quick Start with Helicon Focus (published by HeliconSoft).
- A video tutorial explaining the photo-stitching process can be found at youtube–How to Stitch Multiple Photos Together Automatically with Photomerge in Photoshop (published by Photoshop Video Academy).
- Import each stack of images into Helicon Soft and render focus-stacking.
Notes:- Begin with Method C (pyramid).
- If the edge of the image becomes distorted (halo effect), troubleshoot with Method A (weighted average) or Method B (depth map)–and increase the “radius” value. Read more about the different methods here: https://www.heliconsoft.com/helicon-focus-main-parameters/.
- Other focus-stacking software (e.g., Adobe Photoshop) may be used, though the processing speed and quality of results may vary.
- Repeat for each stack of images. Save images.
- Open all focus-stacked images in Adobe Photoshop and check for processing artifacts (Figure 4). If artifacts exist, use the “Crop” tool (hotkey: C) to discard the artifact. Save all changes before proceeding.

Figure 4. Example of artifacts created during focal stacking. Artifacts such as blurred areas along the edge of the Z-stack may appear (black arrow) and should be cropped out of the image. - With all focus-stacked images opened in Adobe Photoshop and vetted for artifacts, use the photo-stitching tool (File > Automate > Photomerge …).
- Select “Add Open Files” (all files must be already saved), select “Auto”, then “OK”.
Note: If there are many stacks to stitch together, this step may take a while. If computer crashes, try using a computer with more RAM and stronger processor. - Check for artifacts or unphotographed areas. Save image.
- Open the image containing microscope standard taken back in Step C10. Using the “Rectangle Marquee” tool (hotkey: M) to measure a known width (e.g., 1 mm).
- Use the “Brush” tool (hotkey: B) to fill in the rectangle with a color that contrasts well with the background.
- With the colored rectangle still in selection, copy (Windows hotkey: Ctrl + C; Mac hotkey: Cmd + C) the scale bar and paste (Windows hotkey: Ctrl + V; Mac hotkey: Cmd + V) onto the photo-stitched image of the sample. Save image.
- Calibrating measurement tools for image analysis/ionocyte quantification
Note: A video tutorial explaining this process can be found at Youtube–Set calibration in ImageJ (published by remotelab PloyU).- Open the photo-stitched image in FIJI/ImageJ.
- Using the “magnifying glass” tool or by pushing the “+” key, zoom in on the scale bar.
Note: Hover the mouse cursor over the area you want to zoom in before pushing “+”. You can also push “-” to zoom out. - Using the “straight line” tool, draw a line across the scale bar–holding shift to lock the line at a 90° angle.
- At the top of the window (screen for Mac), click on Analyze > Set Scale. The distance in pixels should already be filled in as that is the line you just drew. Fill in the “Known Distance” and “Unit of Length” (e.g., 1 mm), then click “OK”.
Note: If the same scale bar is being used across multiple samples, then select the box next to “Global”. In doing so, the pixel to mm ratio will be remembered across windows (samples). - To ensure accuracy, open up the microscope standard image previously taken during “Step C10”. Using the “straight line” tool, draw a line across the imaged scale bar and measure (hotkey: M). If done properly, the difference between the imaged scale bar and the “measured” distance should be negligible.
Note: From this point forward, this protocol will regard the sample as a fish specimen. Other samples could be similarly quantified, though some modifications to this protocol may be necessary.
- Surface area estimation
- Using FIJI/ImageJ’s “straight line” tool, measure (hotkey: M) the standard length of the fish larva. Record this value.
Note: The “segmented line” tool (right click (Mac: command + click) on the “straight line” tool) can be used if the fish larva was fixed or placed in a curved position. - With the “Freehand Selection” tool, trace the fish larva to measure (hotkey: M) its surface area. Record this value.
Note: If freehand drawing using a mouse proves difficult, try using a tablet and stylus. - Using the “Freehand Selection” tool, trace regions of interest where ionocyte distribution may differ (in this example we use head, body, and fins) and measure (hotkey: M). Record these values.
- Using FIJI/ImageJ’s “straight line” tool, measure (hotkey: M) the standard length of the fish larva. Record this value.
- Counting cutaneous ionocytes
Notes:- This technique is suitable for smaller fish specimens (< 5 mm standard length) and samples with a non-homogenous distribution of ionocytes.
- A video tutorial explaining this process can be found at Youtube–Count Items in ImageJ (published by Timothy Spier).
- Open FIJI/ImageJ.
- If using ImageJ, be sure the “Cell Counter” plugin has been installed (see Software).
- Open the photo-stitched image in FIJI/ImageJ.
- Go to Plugin > Analyze > Cell Counter > Cell Counter.
- In the Cell Counter window, click “Initialize”, select “Type 1”, and zoom (“+”) in on the area of interest.
- Using your mouse, left click to denote a cell at that location. The count will be updated next to “Type 1” in the Cell Counter window. Record this value.
- To remove an erroneous count, check the box next to “Delete Mode”. Next, click on the point you would like to remove. Uncheck the box to proceed with counting.
- Use the “Save Markers” option in the Cell Counter window to save your work.
- Estimating number of cutaneous ionocytes
Note: This technique is suitable for larger fish specimens with a homogenous ionocyte distribution. Because there is likely species-specific variation in ionocyte distribution, this estimation technique needs to be ground truthed with manual cell counts (Procedure G) before utilizing.- Open the photo-stitched image with scale bar in Adobe Illustrator.
Notes:- To zoom in, use the zoom tool (hotkey: Z) and click.
- To zoom out, use the zoom tool (hotkey: Z), hold ctrl (Mac: hold Cmd) and click.
- Adjust the artboard (hotkey: Shift + O) to fit the photo-stitched image. If the photo-stitched image is larger than the maximum artboard size, scale down the image size by 1) clicking on the image, 2) hovering over a corner anchor point, 3) holding “shift”, and 4) dragging to reduce image size.
- Change the name of this layer to “image”, and lock it. Create four additional layers and arrange them in this order: fin count, body count, head count, grid, image (from top to bottom) (Figure 5).
Notes:- You may have other areas of interest requiring additional layers
- A layer is only editable when it is 1) visible, 2) unlocked, and 3) selected.
- Layer(s) at the top will cover the layer(s) at the bottom.
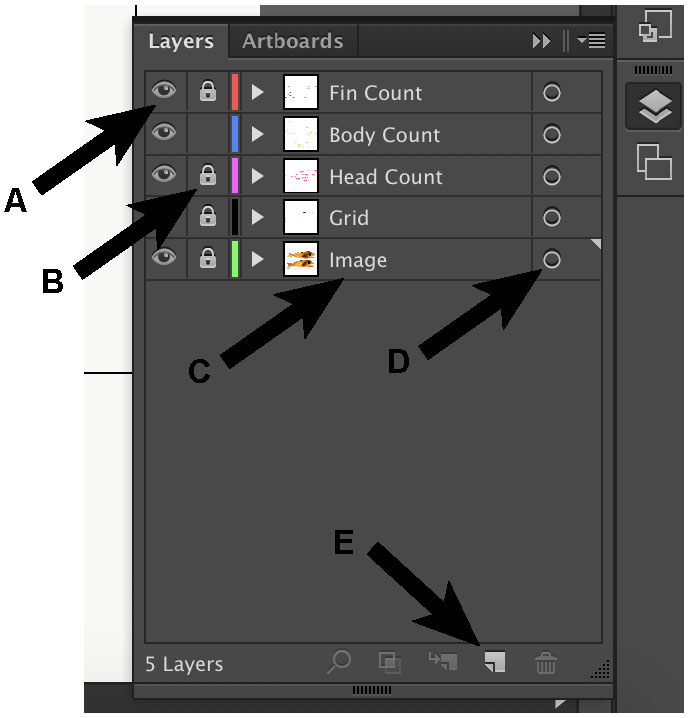
Figure 5. Key buttons within the layers window. The eye symbol (A) controls the visibility of that layer, whereas the lock symbol (B) controls whether that layer is editable. To change the name of the layer, double click on the name (C). To select everything in that layer, click on the circle in the layer of interest (D). To add another layer, click “create new layer” (E). - Toggle the “Ruler” option (Window: Ctrl + R; Mac: Cmd + R). Right click on the ruler and change to “pixels”.
- Using the rectangle tool (hotkey: M), drag a rectangle across the scale bar (Figure 6A).
- On the second row from the top towards the right side of the window, find and click the orange “Transform”.
- Record the value listed next to “W” (Figure 6B). This value will assist you in converting between pixels and the measured metric unit.
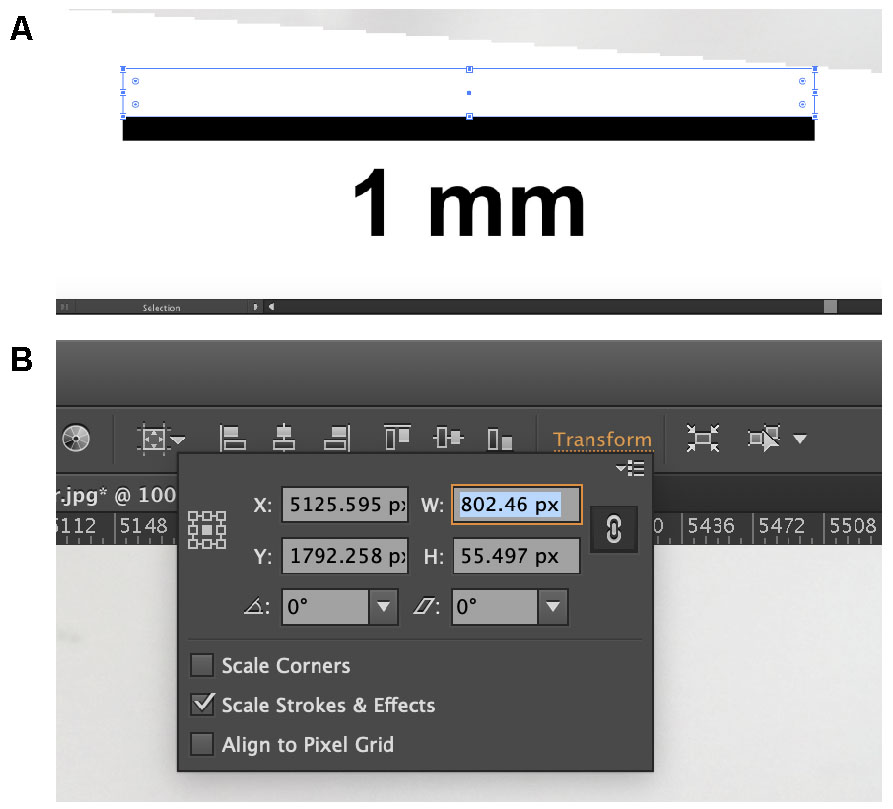
Figure 6. Finding the conversion between digital pixels and measured metric units. Draw a rectangle across the scale bar (A). The width of the rectangle should then appear in the “Transform” window (B). Use this value to convert between the computer pixels and the metric system. - Using the standard length that was calculated back in Step F1, calculate the value equal to 2% of the fish specimen’s standard length.
- Using the rectangle tool, click (not drag) to create a shape with custom width and height dimensions. Input the 2% standard length value into both the “Width” and “Height” dimension (make a square).
Note: For example, if the standard length of specimen = 8.80 mm, then 2% of the standard length is 0.176 mm. - Copy and paste the squares until a grid overlays the entire fish (Figure 7).
Note: To expedite this process, below are some helpful shortcuts:- Select All: “Windows: Ctrl + A, Mac: Cmd + A”
- Copy: “Windows: Ctrl + C; Mac: Cmd + C”
- Paste in place: “Windows: Ctrl + Shift + V; Mac: Cmd + Shift + V”
- While dragging newly pasted cells, hold “Shift” to lock the squares in the X or Y plane.
- By default, illustrator should “auto-snap” your selection to place. If this is turned off, go to Preferences > Selection and Anchor Display and check the box “Snap to Point”.
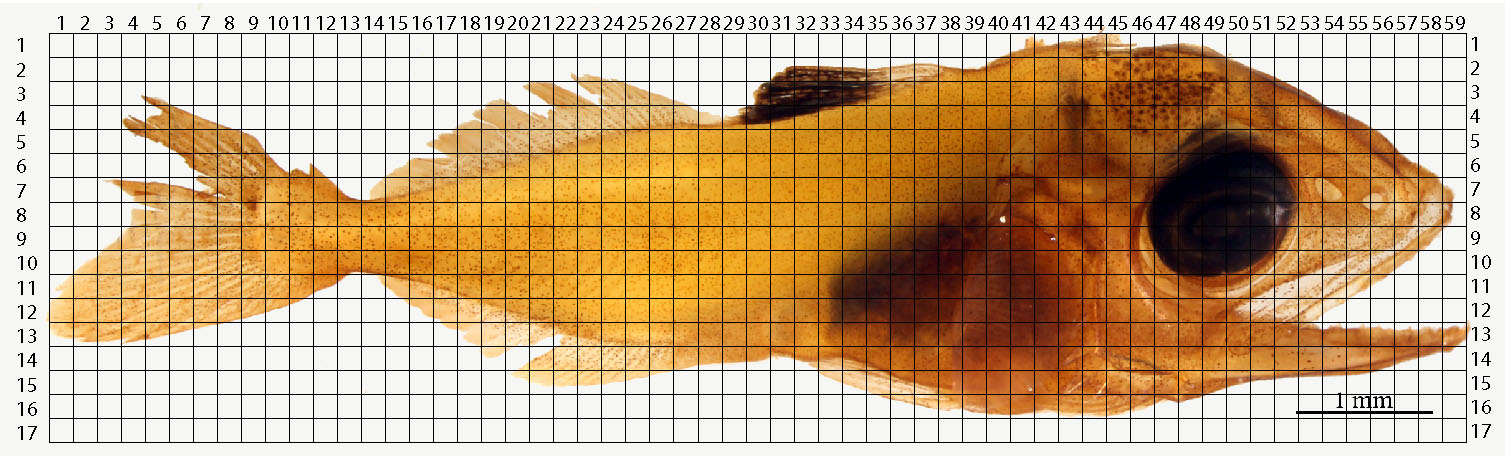
Figure 7. Example of an overlying grid over a photo-stitched yellowfin tuna specimen. Each square’s length is equal to 2% of the larva’s standard length. - Number the grid’s x and y axes.
- Calculate the area of an individual square. Record this value.
Note: For example, if the standard length of specimen = 8.80 mm, then the area of each square is 0.031 mm2. - Using the measurements from Steps F2 and F3, calculate how many squares will require counting to cover 10% of each region of interest (e.g., head, body, fins). Record the number of squares necessary to cover at least 10% of each region.
Notes:- If the area of the head region = 4.9 mm2 and each square is 0.031 mm2, then:
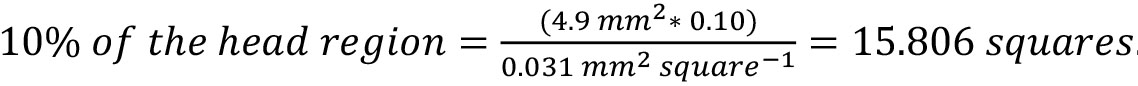 . Round up and count 16 squares to adequately cover at least 10% of the head region.
. Round up and count 16 squares to adequately cover at least 10% of the head region. - Separation of different body regions is necessary as variation in regional ionocyte density may occur due to species-specific or developmental stage-specific differences.
- If the area of the head region = 4.9 mm2 and each square is 0.031 mm2, then:
- Open R. Copy and paste in the provided codes (see Note #1 below). Adjust the number for the X and Y coordinates accordingly before using.
Note: Step-by-step instruction for using this R below:
### Note #1
### Beginning of code
# Generating random coordinates
# Step-by-step instruction
#
# Objective: this will randomly generate 500 (x,y) coordinates
# Step 1) In line 15, replace "59" with the number of squares on the x-axis
# Step 2) In line 16, replace "17" with the number of squares on the y-axis
# Step 3) highlight everything (Windows: ctrl + A; Mac: cmd + A)
# Step 4) hold "Shift" and hit "enter"
# Step 5) see output in the R Console window.
# Note 1: Some coordinates may be repeated; skip if repeated.
# Note 2: If 500 (x,y) coordinates is not enough, repeat until the quota for each region is reached.
x.coord = sample(1:59, replace=T, 500)
y.coord = sample(1:17, replace=T, 500)
rand.coords <- data.frame(x.coord,y.coord)
rand.coords
### End of code - For each randomly generated coordinate, disqualify for counting if the square contains any of the following:
- Background space, glare, or other photography artifacts (Figure 8A).
- Folding, invaginations, pigments, which prevent accurate counts (Figure 8B).
- More than one region of interest: head, body, fins (Figure 8C).
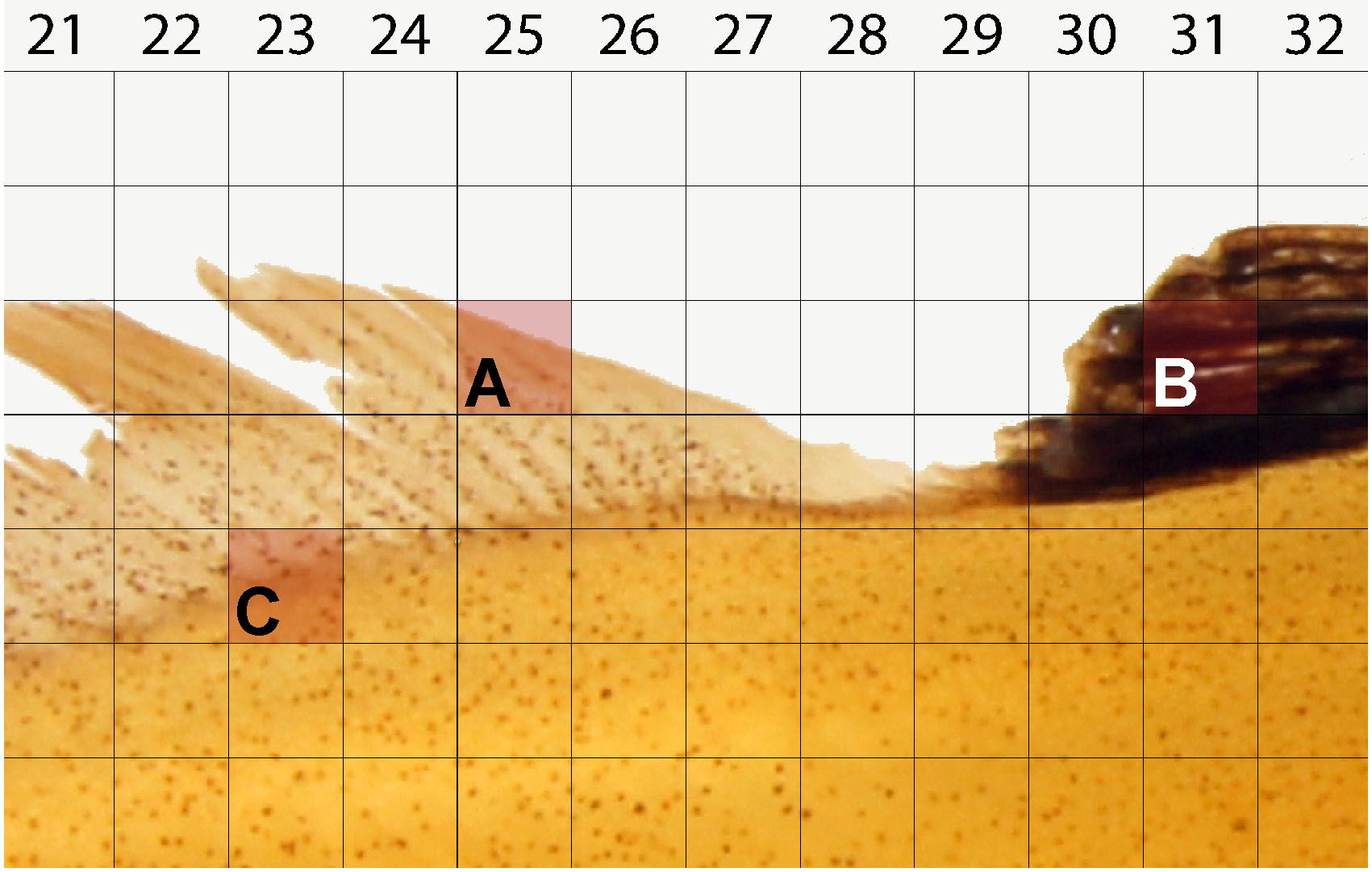
Figure 8. Example of squares unsuitable for cell counting. Randomly generated coordinates must not contain the following features: background space (A), foldings, invaginations, pigments, or other dark space (B), and space inclusive of more than one region e.g., box containing both body and fin (C).
- If the square meets the above criteria, then mark for counting.
Notes:- To quickly designate the square for counting, use the “Pencil tool” (hotkey: N) to scribble a marking.
- To avoid accidently moving the grid or the underlying image, be sure to lock both the grid and image layer (see Figure 5B).
- Count the cells within Adobe illustrator. For each region, record the number of ionocytes present in the randomly-chosen grid squares.
Note: To quickly record the number of cells in the square, use the “Type tool” (hotkey: T) and click within the square, then type the number of counted cells. Hit “Esc” to exit this mode. - If the grid lines happen to overlap the immunostained cells, count only the cells intersecting with the top and right sides of the square. This will ensure no double-counting of the cells if two squares were randomly chosen next to each other.
Note: If the grid lines are too thick or the cells are located directly underneath the lines, try selecting the grid layer (hit the rightmost circle in the grid layer; Figure 5D) and reducing the “stroke” value to “0.5”, or reducing the opacity to 50% for better viewing. - Continue until at least 10% of each region (head, body, fins) have been counted (Figure 9).
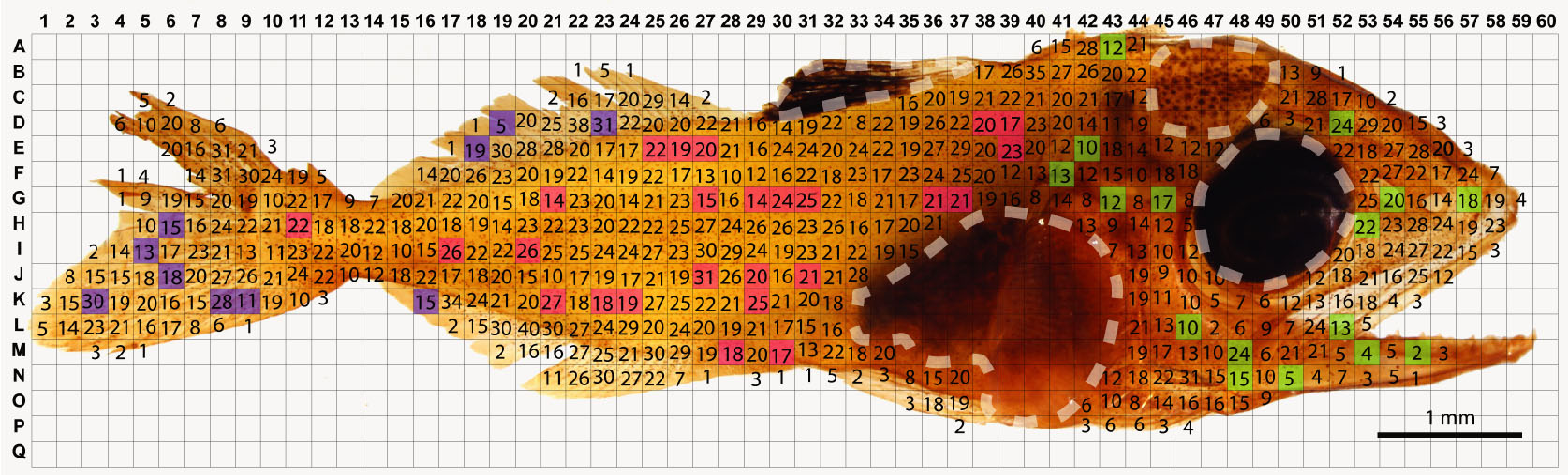
Figure 9. Method for quantification of cutaneous ionocytes in yellowfin tuna specimen and early stage juveniles (> 5 mm standard length). Ionocytes were identified by their intense Na+/K+-ATPase immunostaining and counted within randomly sampled boxes of the overlaid grid within the head (green), body (pink), and fin (blue) regions. Dashed white lines outline regions that were not sampled due to heavy pigmentation preventing accurate ionocyte counts. The estimated ionocyte count (15,342 cells) was ground truthed with manual ionocyte count (colorless regions; 14,795 cells). The estimated relative ionocyte area for this larva is 3.75%. Figure from Kwan et al., 2019. - Calculate the estimation using the following equation:
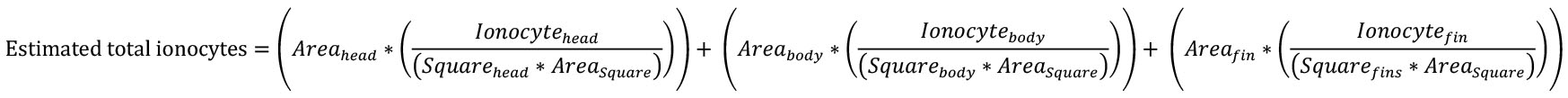
Notes:- Areahead, body, or fins = measured area of head, body, or fins region (Step F3).
- Ionocytehead, body, or fins = sum of ionocytes counted per region (Step H17).
- Squarehead, body, or fins = number of squares counted per region (Step H13).
- Areasquare = size of each square = (standard length*0.02)2 (Step H12).
- Open the photo-stitched image with scale bar in Adobe Illustrator.
- Quantifying ionocyte size
- Open up ImageJ and set scale bar as discussed in Procedure E.
- With the “Freehand Selection” tool, zoom in (hotkey: +) and trace individual cutaneous ionocytes to estimate their relative surface areas.
Note: If freehand drawing proves difficult, try using a tablet and stylus. - Repeat Step I2 until you have measured 20 cells from different regions of interest (e.g., head, body, and fins).
- Calculate the average ionocyte size using the following equation:
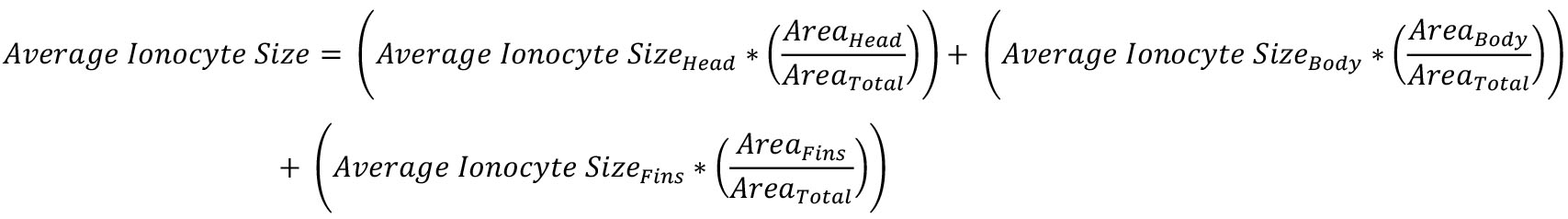
Areahead, body, or fins were calculated in Step F3.
Areatotal was calculated in Step F2.
- Quantifying relative ionocyte area
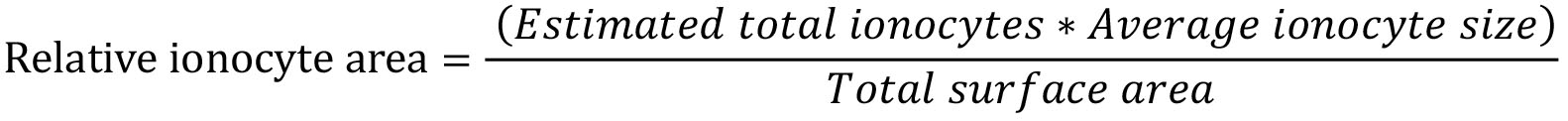
Estimated total ionocytes were calculated in Step H20.
Average ionocyte size were calculated in Step I4.
Total surface area were calculated in Step F2. - Method controls and antibody validation
- Non-specific secondary and tertiary antibody binding control: designate several fish specimens as negative controls by omitting the primary antibody during Step B6 (leave in NHS blocking solution overnight at 4 °C). Fish specimens incubated with the primary NKA antibody should result in positive staining (Figure 10A). In contrast, fish specimens without the primary antibody should result in the absence of staining (Figure 10B).
Notes:- Performing this negative control step is a great way to identify and avoid the natural pigments present on the organism of interest (Step H15).
- If necessary, the negative-control fish specimens can be re-incubated and utilized. Simply repeat all of Procedure B to re-immunostain said sample(s).
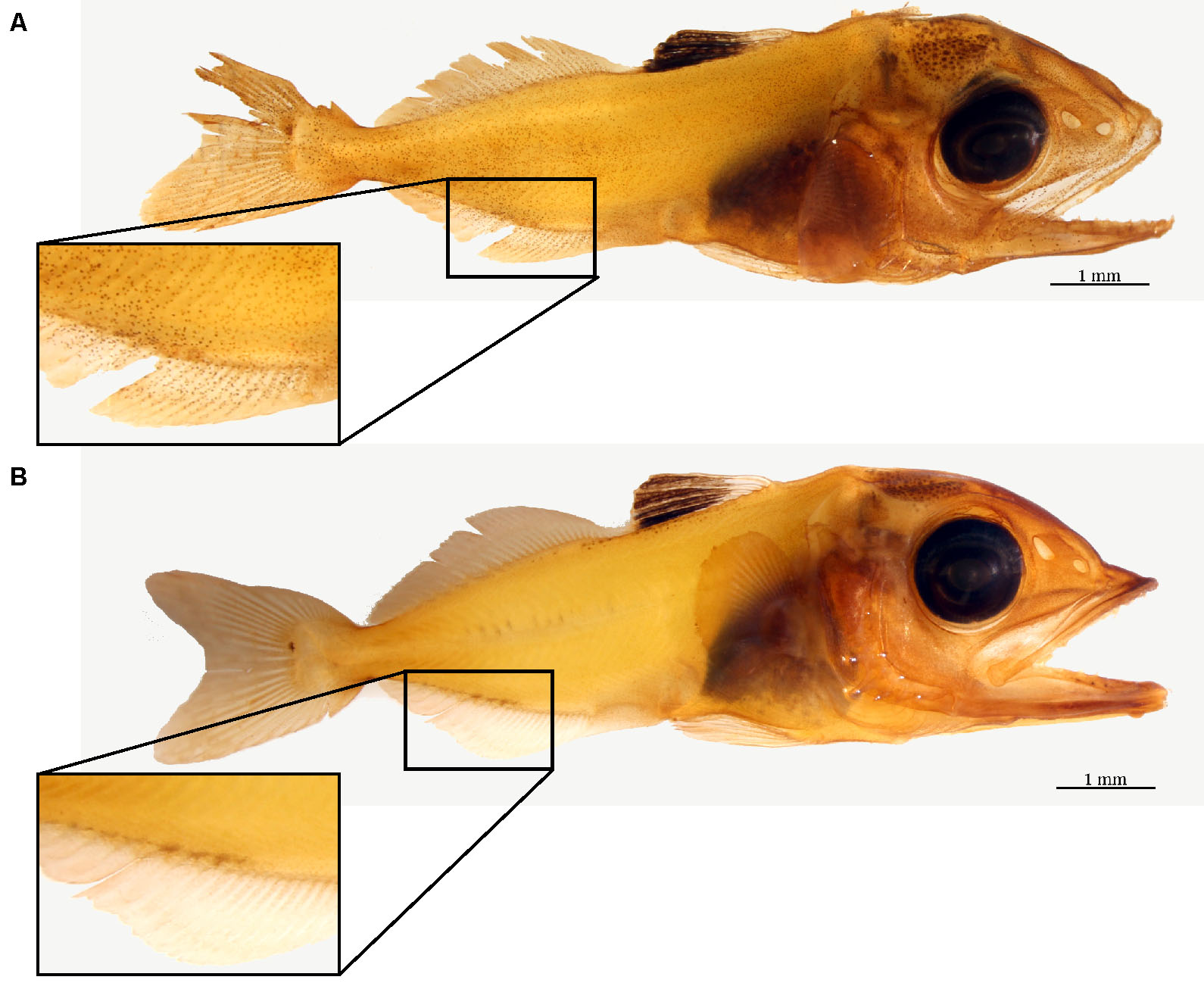
Figure 10. Negative control for immunohistochemistry. The difference between a yellowfin tuna specimen incubated with (A) and without (B) Na+/K+-ATPase (NKA) antibodies. The brown precipitate seen in the top larva denotes NKA presence on the cutaneous surface, whereas the absence of brown staining on the bottom larva serves a successful negative control. Note the areas containing naturally occurring pigment in both specimens should be avoided during estimation. - Primary antibody specificity via western blotting: flash frozen tissue (-80 °C) should be sampled along with the fixed tissue for western blotting, which should yield a single band close the predicted size of your protein of interest (Figure 11). See supplementary material 2 in Kwan et al. (2019) for protocol and additional details.
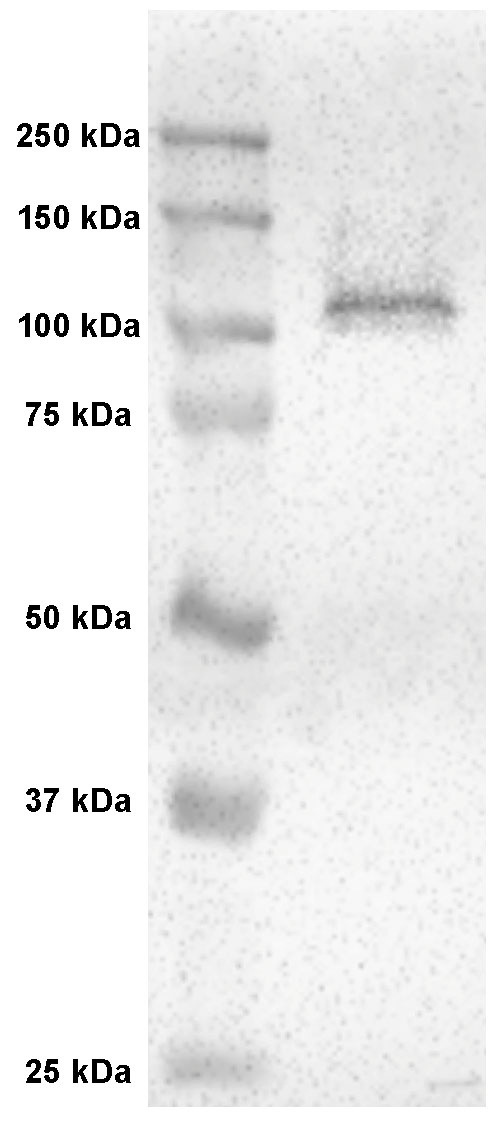
Figure 11. Western blot of Na+/K+-ATPase (NKA) in yellowfin tuna tissue. Western blot with anti-NKA monoclonal antibodies on yellowfin tuna tissue yielded a single ~108 kDa band, which matched the predicted size of the protein. Figure adapted from Kwan et al., 2019.
- Non-specific secondary and tertiary antibody binding control: designate several fish specimens as negative controls by omitting the primary antibody during Step B6 (leave in NHS blocking solution overnight at 4 °C). Fish specimens incubated with the primary NKA antibody should result in positive staining (Figure 10A). In contrast, fish specimens without the primary antibody should result in the absence of staining (Figure 10B).
Data analysis
Ionocyte count (Step H20), average ionocyte size (Step I4), and relative ionocyte area (Step J1) data across development is best depicted in a scatterplot and explained using regression models. In Kwan et al., 2019, we examined the relationships of relative ionocyte area across standard length and days post hatching using linear regressions. The slope of the linear regression explains the steepness of the line, with the null hypothesis being zero, whereas R2 describes the regression’s goodness-of-fit. Other developmental metrics that may be applicable include accumulated thermal units and total length of the sample. Development may not always be linear; therefore, researchers should choose their own regressions according to your field of study. If the experiment includes multiple treatments or species, the comparison of the regression’s slope should be examined with t-test (2 treatments total) or Analysis of Covariance (3 or more treatments). See Zar (2014) for additional details.
Recipes
- 1.2x phosphate buffer saline (PBS) solution (1 L)
120 ml of 10x PBS stock solution
880 ml of deionized water - 4% paraformaldehyde (PFA) fixative solution (10 ml)
a.Break open a PFA ampule (20% stock solution) within the fume hood
b.Add 2 ml of PFA stock solution into 8 ml of 1.2x PBS solution (Recipe 1), for a final concentration of 4% PFA in 1 M PBS. Invert to mix
c.Remaining PFA stock solution can be stored at 4 °C for up to six months
d.4% PFA fixative solution can be stored at 4 °C for up to one week - 3% hydrogen peroxide solution (10 ml)
1 ml of 30% hydrogen peroxide
9 ml of tap water - 1.0x PBS solution (1 L)
100 ml of 10x PBS stock solution
900 ml of deionized water - DAB substrate working solution (same as manufacturer instruction) (~5 ml)
5 ml of DI water
2 drops of buffer stock solution (DAB substrate kit)
4 drops of DAB stock solution (DAB substrate kit)
2 drops of hydrogen peroxide solution (DAB substrate kit) - 10% sodium azide stock solution (10 ml)
10 ml of 1.0x PBS
1 g of sodium azide - 0.02% sodium azide working solution (10 ml)
10 ml 1.0x PBS
20 μl of 10% sodium azide stock solution (Recipe 6)
Acknowledgments
This protocol corresponds to the methods and experiments presented in Kwan et al. (2019). The larval yellowfin tuna and white seabass samples (Atractoscion nobilis) used to create and optimize this protocol were provided by the Inter-American Tropical Tuna Commission (IATTC) and the Hubbs Sea World Research Institute (HSWRI). We are grateful to Jeanne Wexler (IATTC) for her contributions to the original project, and for rearing and sampling the yellowfin tuna larvae. We thank Diomedes Ballesteros, Lina Castillo, Susana Cusatti, Danisin Dominguez, Agustin Ortega, Daniel Perez, Dario Ramires, Daniel Solis, Luis Tejada, and Carlos Vergara for assisting with the production and rearing of yellowfin tuna larvae at the Achotines Laboratory in Panama. We thank Daniel Margulies (IATTC) and Vernon Scholey (IATTC) for developing the yellowfin tuna rearing methods and supervising the spawning and larval rearing at the Achotines Laboratory. We are grateful to Erica Brombay-Fanning (HSWRI), Eric McIntire (HSWRI), Sabrina Sobel (HSWRI), and Christy Varga (HSWRI) for the rearing and sampling the white seabass larvae of larval white seabass. We thank Mark Drawbridge (HSWRI) for supervising the spawning and larval rearing at the Leon R. Raymond Marine Fish Hatchery in Carlsbad, USA. The authors would like to thank Dr. Greg Rouse for the use of microscope and camera equipment, Sabine Faulhaber for technical assistance with the scanning electron microscope, and Taylor Smith for her assistance in dissection and imaging. G.T.K. was supported by the San Diego Fellowship and the National Science Foundation Graduate Research Fellowship Program.
Competing interests
The authors declare no competing interests.
Ethics
All experiments were approved by the Scripps Institution of Oceanography/University of California, San Diego animal care committee under protocol no. S10320 in compliance with the Institutional Animal Care and Use Committee (IACUC) guidelines for the care and use of experimental animals.
References
- Clifford, A. M., Goss, G. G., Roa, J. N. and Tresguerres, M. (2015). Acid/base and ionic regulation in hagfish. In: Hagfish Biology. Science Publishers, Boca Raton, pp. 277-292.
- Evans, D. H., Piermarini, P. M. and Choe, K. P. (2005). The multifunctional fish gill: dominant site of gas exchange, osmoregulation, acid-base regulation, and excretion of nitrogenous waste. Physiol Rev 85(1): 97-177.
- Fridman, S., Bron, J. E. and Rana, K. J. (2011). Ontogenetic changes in location and morphology of chloride cells during early life stages of the Nile tilapia Oreochromis niloticus adapted to fresh and brackish water. J Fish Biol 79(3): 597-614.
- Hiroi, J., Kaneko, T., Seikai, T. and Tanaka, M. (1998). Developmental sequence of chloride cells in the body skin and gills of Japanese flounder (Paralichthys olivaceus) larvae. Zoolog Sci 15(4): 455-460.
- Hiroi, J., Kaneko, T. and Tanaka, M. (1999). In vivo sequential changes in chloride cell morphology in the yolk-sac membrane of mozambique tilapia (Oreochromis mossambicus) embryos and larvae during seawater adaptation. J Exp Biol 202 Pt 24: 3485-3495.
- Hwang, P. P. (1989). Distribution of chloride cells in teleost larvae. J Morphol 200(1): 1-8.
- Katoh, F., Shimizu, A., Uchida, K. and Kaneko, T. (2000). Shift of chloride cell distribution during early life stages in seawater-adapted Killifish, Fundulus heteroclitus. Zoolog Sci 17(1): 11-18.
- Kwan, G. T., Wexler, J. B., Wegner, N. C. and Tresguerres, M. (2019). Ontogenetic changes in cutaneous and branchial ionocytes and morphology in yellowfin tuna (Thunnus albacares) larvae. J Comp Physiol B 189(1): 81-95.
- Piermarini, P. M. and Evans, D. H. (2001). Immunochemical analysis of the vacuolar proton-ATPase B-subunit in the gills of a euryhaline stingray (Dasyatis sabina): effects of salinity and relation to Na+/K+-ATPase. J Exp Biol 204(Pt 19): 3251-3259.
- Roa, J. N., Munevar, C. L. and Tresguerres, M. (2014). Feeding induces translocation of vacuolar proton ATPase and pendrin to the membrane of leopard shark (Triakis semifasciata) mitochondrion-rich gill cells. Comp Biochem Physiol A Mol Integr Physiol 174: 29-37.
- Roa, J. N. and Tresguerres, M. (2017). Bicarbonate-sensing soluble adenylyl cyclase is present in the cell cytoplasm and nucleus of multiple shark tissues. Physiol Rep 5(2). pii: e13090.
- Tang, C. H., Leu, M. Y., Yang, W. K. and Tsai, S. C. (2014). Exploration of the mechanisms of protein quality control and osmoregulation in gills of Chromis viridis in response to reduced salinity. Fish Physiol Biochem 40(5): 1533-1546.
- Varsamos, S., Diaz, J. P., Charmantier, G., Blasco, C., Connes, R. and Flik, G. (2002). Location and morphology of chloride cells during the post-embryonic development of the european sea bass, Dicentrarchus labrax. Anat Embryol (Berl) 205(3): 203-213.
- Varsamos, S., Nebel, C. and Charmantier, G. (2005). Ontogeny of osmoregulation in postembryonic fish: a review. Comp Biochem Physiol A Mol Integr Physiol 141(4): 401-429.
- Wilson, J. M., Randall, D. J., Donowitz, M., Vogl, A. W. and Ip, A. K. (2000). Immunolocalization of ion-transport proteins to branchial epithelium mitochondria-rich cells in the mudskipper (Periophthalmodon schlosseri). J Exp Biol 203(Pt 15): 2297-2310.
- Wilson, J. M., Whiteley, N. M. and Randall, D. J. (2002). Ionoregulatory changes in the gill epithelia of coho salmon during seawater acclimation. Physiol Biochem Zool 75(3): 237-249.
- Yang, W. K., Kang, C. K., Chang, C. H., Hsu, A. D., Lee, T. H. and Hwang, P. P. (2013). Expression profiles of branchial FXYD proteins in the brackish medaka Oryzias dancena: a potential saltwater fish model for studies of osmoregulation. PLoS One 8(1): e55470.
- Zar, J.H. (2014). Comparing simple linear regression. In: Biostatistical Analysis. In: Pearson, H. (Ed.). pp: 387-404.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kwan, G. T., Finnerty, S. H., Wegner, N. C. and Tresguerres, M. (2019). Quantification of Cutaneous Ionocytes in Small Aquatic Organisms. Bio-protocol 9(9): e3227. DOI: 10.21769/BioProtoc.3227.
Category
Cell Biology > Cell imaging > Fixed-tissue imaging
Biochemistry > Protein > Immunodetection > Immunostaining
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.