- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Dual Fluorescence Reporter Based Analytical Flow Cytometry for miRNA Induced Regulation in Mammalian Cells

.jpg)
Published: Vol 8, Iss 17, Sep 5, 2018 DOI: 10.21769/BioProtoc.3000 Views: 12734
Reviewed by: HongLok LungYi CuiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
MicroRNA-induced gene regulation is a growing field in basic and translational research. Examining this regulation directly in cells is necessary to validate high-throughput data originated from RNA sequencing technologies. For this several studies employ luciferase-based reporters that usually measure the whole cell population, which comes with low resolution for the complexity of the miRNA-induced regulation. Here, we provide a protocol using a dual-fluorescence reporter and flow cytometry reaching single cell resolution; the protocol contains a simplified workflow that includes: vector generation, data acquisition, processing, and analysis using the R environment. Our protocol enables high-resolution measurements of miRNA induced post-transcriptional gene regulation and combined with system biology it can be used to estimate miRNAs proficiency.
Keywords: miRNABackground
MicroRNAs (miRNA) are highly conserved small non-protein-coding RNAs (21-22 nt) that regulate post-transcriptionally gene expression and modulate fundamental biological processes like development and cell homeostasis ( Lagos-Quintana et al., 2001; Fabian et al., 2010; Bartel, 2018), including as well several pathologies where miRNA expression correlates with tumor progression and aggressiveness (Lu et al., 2005; Di Leva and Croce, 2013; Krishnan et al., 2015; Bertoli et al., 2015 and 2016). The study of miRNAs has boosted the evolution of numerous biochemical and computational techniques that unravel new mechanisms and networks applicable to clinical scenarios. These high-throughput procedures include profiling methods (microarrays and NGS), in vivo target validation and single-molecule imaging (Steinkraus et al., 2016). In contrast, functional reporters that directly assess miRNA-target repression have been kept simple and with low resolution.
Deep sequencing and mass spectrometry have increased the possibilities to assess miRNAs and targets expression (Ender et al., 2008; Yang et al., 2009; Brameier et al., 2011; Schramedei et al., 2011; Bai et al., 2014a and 2014b; Muller et al., 2015; Yu et al., 2015), since quantifying the number of RNA molecules and running statistical analyses for the differential expression (miRNAs or mRNA) can generate hypotheses that need further experimental evaluation, using, for example, miRNA exogenous expression or inhibition. In brief mRNA analysis on cDNA level (qPCR or next-generation sequencing) estimates RNAs degradation, mass spectrometry defines the affected coding targets (Yang et al., 2009), and ribosome profiling determines which miRNAs targets are translationally repressed (Bazzini et al., 2012).
Moreover, to narrow down the analysis to the Argonaute bound miRNA captured-based profiling has been used, these profiles rely on RNA isolation from ribonucleoproteins, cDNA synthesis and its quantification (qPCR, microarrays or NGS). Crosslink-immunoprecipitation (CLIP) together with high-throughput sequencing increase enormously our understanding of miRNA regulation (Hafner et al., 2010), to the extent that including an RNA-RNA ligation step (in the capture) allows detecting miRNA-mRNA hybrids called chimeras (Helwak et al., 2013; Grosswendt et al., 2014; Moore et al., 2015).
FRET-based assays in vitro have calculated kinetic parameters for AGO-miRNA binding and RISC formation, increasing the structural basis of miRNA target recognition and suggesting that AGO-miRNA behaves more like RNA binding domain rather than RNA-RNA solely interaction (Wee et al., 2012; Salomon et al., 2015). Using this vast amount of information computational scientist developed algorithms to understand miRNA as an intricate network (Vera et al., 2013; Lai et al., 2016), expanding further with these integrative models the concepts of miRNA as fine-tuners and switches of gene expression (Bartel and Chen, 2004).
The increased resolution of sequencing methods did not boost the development of better in vivo reporters. Usually, studies involved in exhaustive biochemical characterizations (Bait profiling) tested functionally miRNA-mRNA interaction using luciferase reporter assays (Helwak et al., 2013; Hasler et al., 2016; Steinkraus et al., 2016). These experiments require two proteins (a reference protein and another with a miRNA response element) to create quantitative results (ratios) for miRNA regulation, dismissing the intricate functional network behind miRNAs; creating a misbalance between the high-resolution biochemical outputs (NGS, CLIP-Seq, FRET, etc.) and low-resolution reporters.
For that reason, we implemented a system for analytical flow cytometry (Denzler et al., 2016), using a single plasmid reporter system and validated it for miRNAs (Lemus-Diaz et al., 2017). To increase resolution, we used fluorescent proteins instead of luciferases, analyzed single cells by flow cytometry, and processed the data using the R environment. Furthermore, we adopted a titration model for miRNAs regulation, tested and validated its prediction, creating three categorical variables that integrate miRNA binding and expression (Mukherji et al., 2011; Lemus-Diaz et al., 2017), which can be used to estimate miRNA proficiency (Garcia et al., 2011).
Here, we provide a detailed protocol that includes plasmid generation, transfection, data acquisition, data handling and plotting using a simplified code. The reporter described here expresses two fluorescent proteins (CFP and YFP) under control of two constitutive promoters (Figure 1A), while one protein is the reference (YFP); the other has a miRNA response element (CFP). In the empty plasmid (CFP w/o miRNA target site) the two fluorescent proteins are expressed proportionally (Figure 1B), while in a plasmid with a miRNA response element this proportionality is shifted (Figure 1C).
To tidy up the raw events and analyze them using a threshold model for miRNA regulation, we process the data into a transfer function (called here Analytical function) by binning the reference protein intensities and calculating the mean for sensor protein (Bosson et al., 2014; Denzler et al., 2016). For our construct, we transfer the raw data in FCS 2.0 format into the R environment using FlowCore Bioconductor package (Gentleman et al., 2004; Hahne et al., 2009; Huber et al., 2015), then we logarithmically transform the YFP relative intensities, bin them at 0.05 intervals, and calculate the average of the log CFP intensities of each range.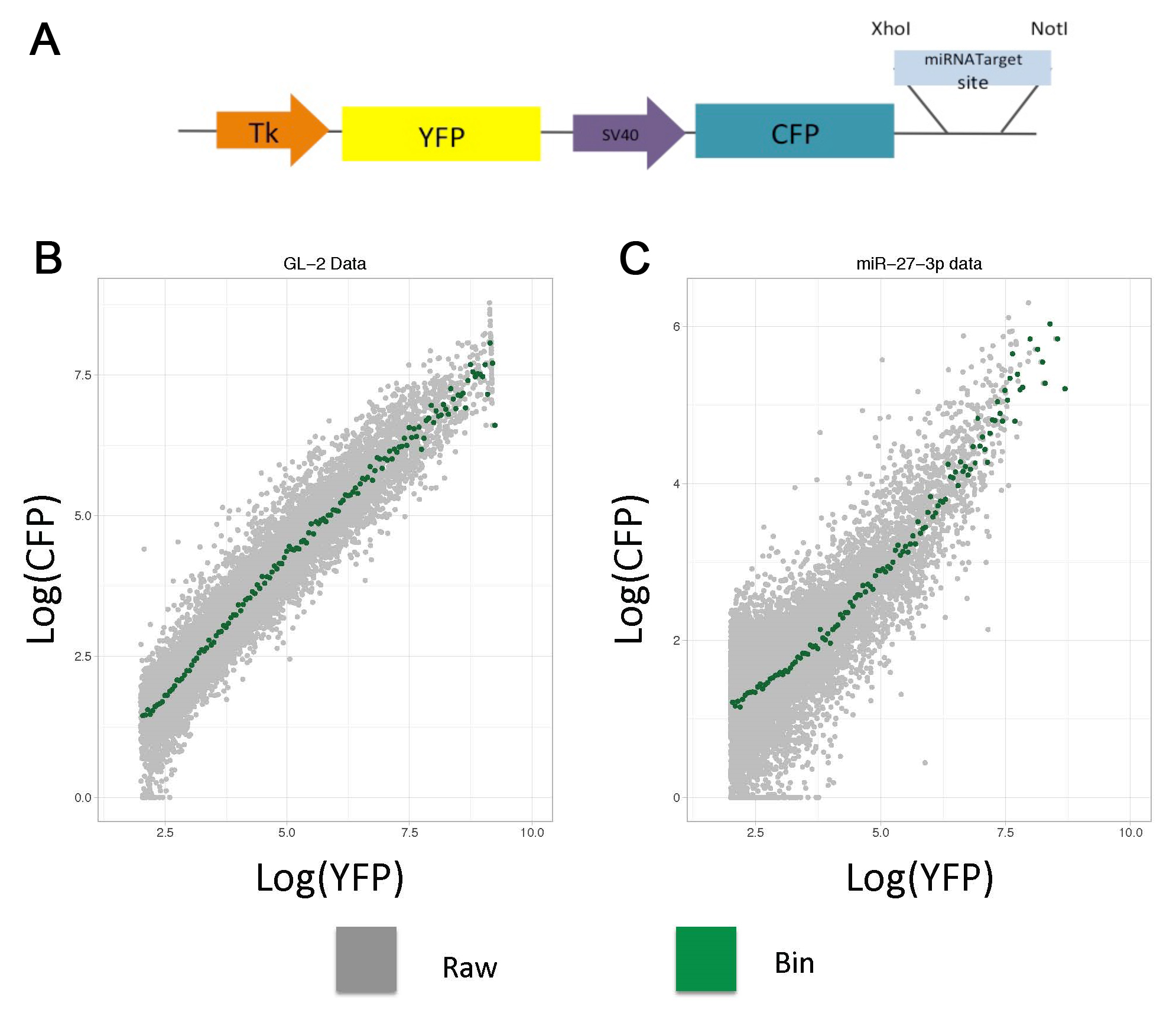
Figure 1. Dual fluorescence-reporter analytical flow cytometry. A. The plasmid contains two fluorescent proteins (YFP and CFP) with two constitutive promoters and a miRNA response element. B and C. To generate analytical functions using raw cytometry data (Grey dots), the YFP relative intensities are binned at 0.05 intervals, and the average CFP intensity per bin is calculated (Green dots). Transfection of HEK 293 with two constructs: (B) Non-cognate and (C) miR-27-3p targeted insert.
Materials and Reagents
- Plasmids, primers and cells
- Other materials
- Pipette tips
- 1,000 μl Filter Tips (SARSTEDT, catalog number: 70.762.211 )
- 100 μl Filter Tips (SARSTEDT, catalog number: 70.760.212 )
- 10 μl Filter Tips (SARSTEDT, catalog number: 70.1116.210 )
- 1.5 ml microfuge tubes (Eppendorf, catalog number: G_0030108116 )
- 2.0 ml microfuge tubes (SARSTEDT, catalog number: 72.695.200 )
- 24-well plates (Greiner Bio One International, catalog number: 662160 )
- 5 ml Ploystyrene Round-Bottom Tube (Corning, Falcon®, catalog number: 352052 )
- PCR tubes 0.2 ml (SARSTEDT, catalog number: 72.737.002 )
- Parafilm (Sigma-Aldrich, Bemis, catalog number: P7793-1EA )
- HEK293 Human embryonic kidney cell line (ATCC, catalog number: CRL-1573TM )
- One Shot® TOP10 Chemically Competent Escherichia coli (Thermo Fisher Scientific, catalog number: C404010 )
- Sense and antisense strands of oligonucleotides (more information below)
- Gel extraction kit (QIAquick gel extraction kit, QIAGEN, catalog number: 28704 )
- GibcoTM Opti-MEMTM (+ L-Glutamine, + phenol red) (Thermo Fisher Scientific, catalog number: 31985047 )
- Lipofectamine® 2000 (Thermo Fisher Scientific, catalog number: 11668027 )
- NotI restriction enzyme (New England Biolabs, catalog number: R0189S )
- XhoI restriction enzyme (New England Biolabs, catalog number: R0146S )
- EcoRI (New England Biolabs, catalog number: R0101S )
- HindIII (New England Biolabs, catalog number: R0104S )
- Plasmid DNA Midiprep kit (QIAquick Plasmid Midi Kit, QIAGEN, catalog number: 10023 )
- QIAprep Spin Miniprep Plasmid mini kit (QIAGEN, catalog number: 27104 )
- Paraformaldehyde 4% solution (Santa Cruz Biotechnology, catalog number: sc-281692 )
- T4 DNA ligase kit (New England Biolabs, catalog number: M0202S )
- T4 Polynucleotide kinase (PNK) (New England Biolabs, catalog number: M0201S )
- Agarose (Carl Roth, catalog number: 3810.3 )
- PBS (PAN-Biotech, catalog number: P04-36500 )
- Trypsin-EDTA (PAN-Biotech, catalog number: P10-023100 )
- DMEM (Thermo Fisher Scientific, Life Technologies, catalog number: 41965062 )
- Fetal Bovine Serum (Thermo Fisher Scientific, Life Technologies, catalog number: 10500-064 )
- Pipette tips
Equipment
- Pipettes
- Fridge
- Agarose gel electrophoresis chamber
- BD LSR II Flow Cytometer (BD, model: LSR II )
- YFP: Laser 488 nm and 550LP-BP575/26 filters
- CFP: Laser 408 nm and BP450/50 (Pacific Blue)
- Benchtop refrigerated microcentrifuge (Thermo Fisher Scientific, model: HeraeusTM FrescoTM 21 , catalog number: 75002555)
- Universal Centrifuge (Thermo Fisher Scientific, model: HeraeusTM MegafugeTM 16 , catalog number: 75004230; Rotor: Thermo Fisher Scientific, model: TX-150, catalog number: 75005701 )
- Gel documentation system (INTAS Gel iX Imager) (INTAS Science Imaging Instruments, model: FACE )
- NanoDropTM 2000 (Thermo Fisher Scientific, model: NanoDropTM 2000 , catalog number: ND-2000)
- Thermoblock (Eppendorf, model: ThermoStat Plus , catalog number: 5352 000.010)
- Thermocycler (Labcycler Sensoquest)
- Vortexer (Scientific Industries, model: Vortex-Genie 2 , catalog number: SI-0236)
Software
- BD-FACSDIVA (BD Biosciences)
- R (https://www.r-project.org)
- R studio (https://www.rstudio.com)
- Bioconductor (https://www.bioconductor.org)
- Serial Cloner (http://serialbasics.free.fr/Serial_Cloner.html)
Procedure
This protocol describes two blocks (Figure 2), one specifies how to generate a reporter plasmids with a miRNA response element, while the second outlines data collection from FACS and processing in the R environment, the second block includes the gates used and a simplified code to generate analytical functions.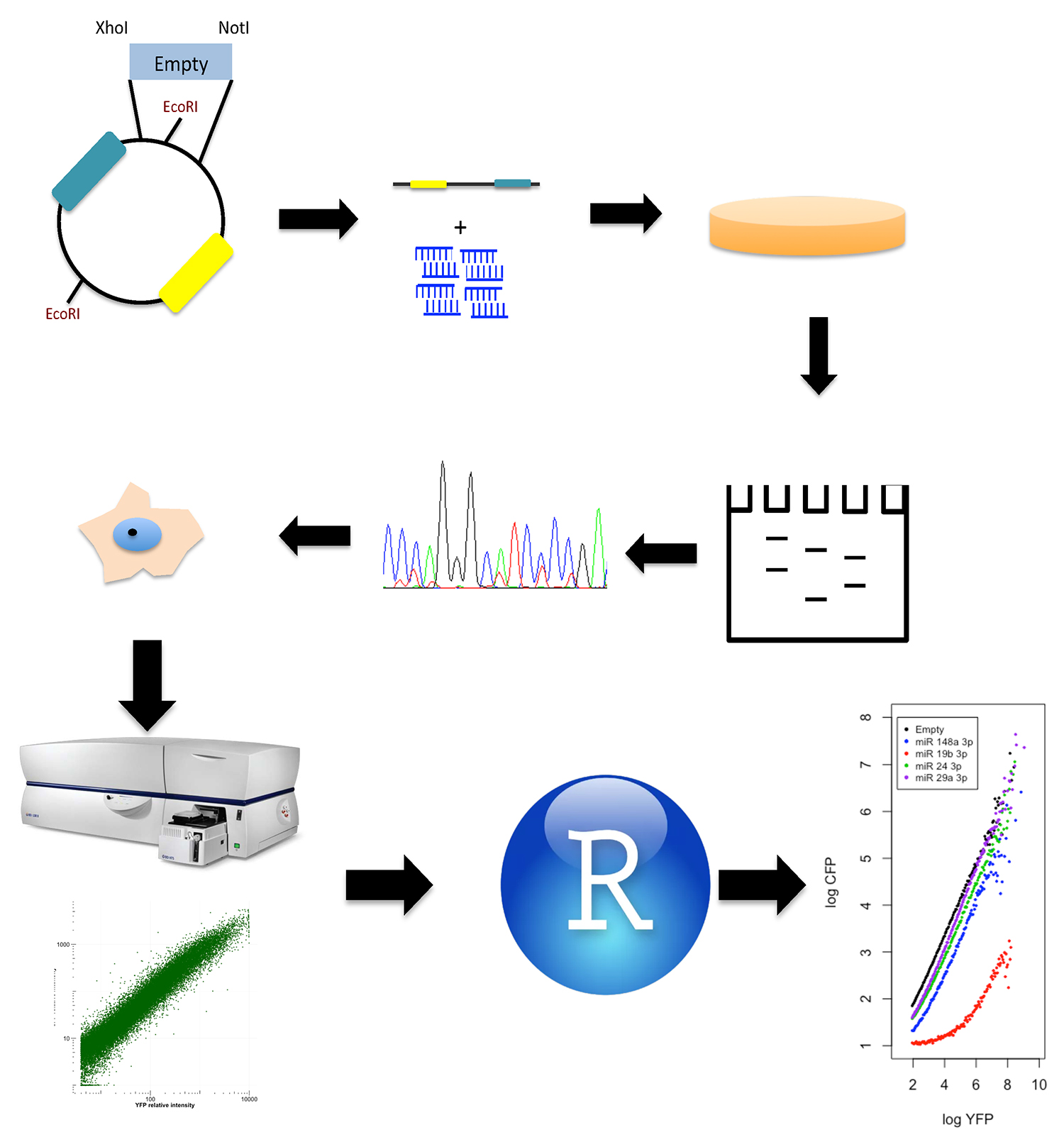
Figure 2. Procedure scheme. Dual reporter plasmid is linearized with XhoI and NotI and ligated with annealed oligonucleotides. Proper oligonucleotides ligation eliminates one of two EcoRI restriction sites in the plasmid; hence positive clones are screened using endonuclease restriction. After the clones are sequenced and transfected, the fluorescence intensities are measured by Flow Cytometry, and the raw data is processed using the R programing language to generate analytical functions.
- Oligonucleotide designing and annealing
Here, we describe how to make a p.UTA.2.0 plasmid with 1 microRNA-binding site, using miR-451a as an example.
Note: We use here perfect miRNA complementarity, but endogenous 3’ UTR can be used.- Design and order oligonucleotides as separate strands (Our oligonucleotides are ordered from Sigma-Aldrich) (Table 1).
Table 1. Oligonucleotides design with XhoI and NotI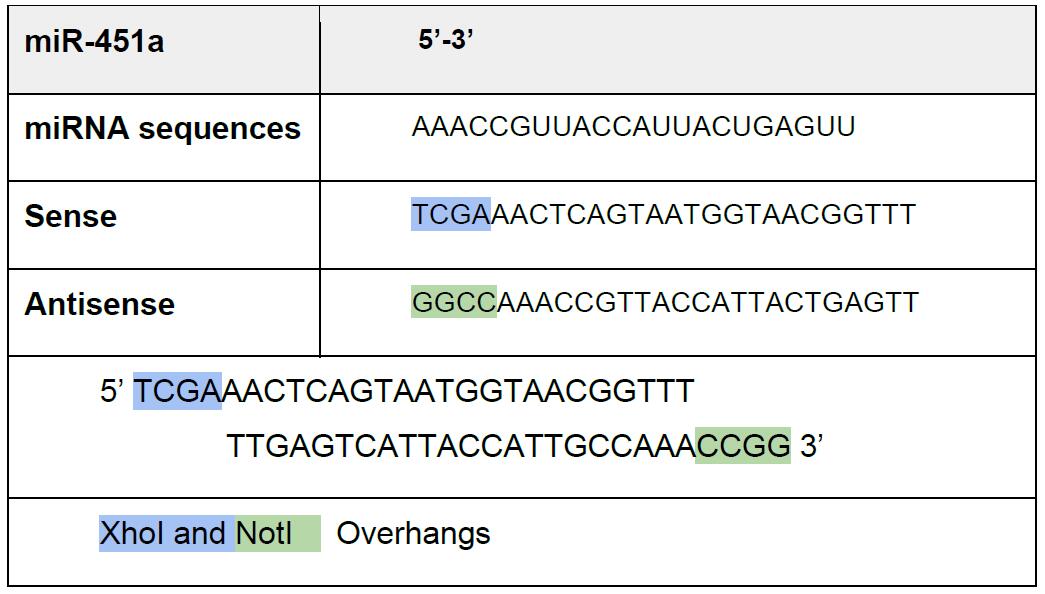
- Prepare in 0.2 ml PCR tubes the following reaction to anneal the oligonucleotides (Table 2).
Table 2. Oligonucleotides annealing and phosphorylation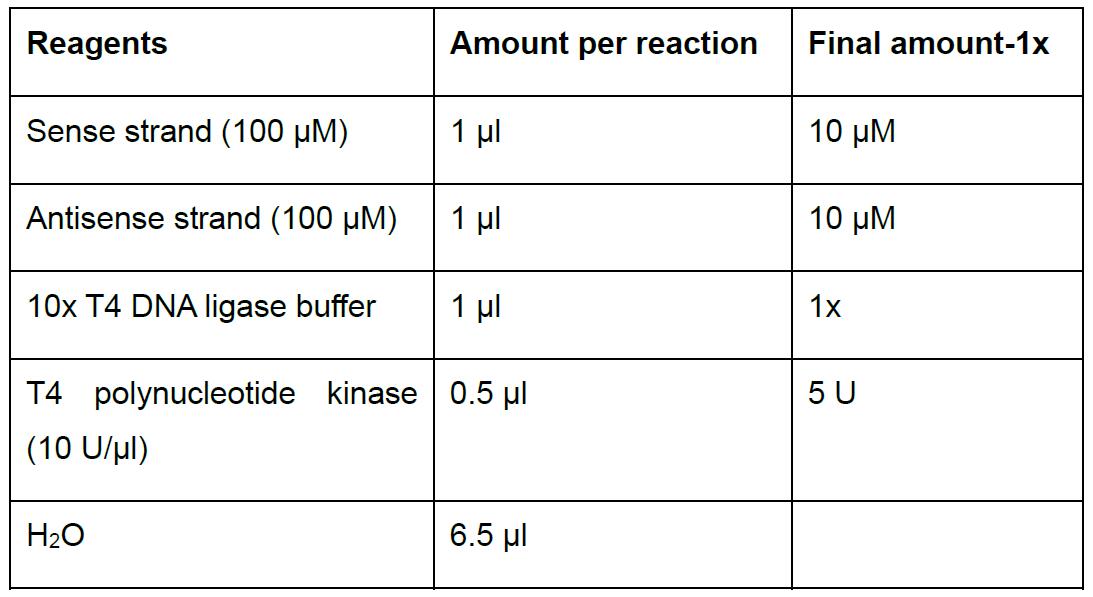
- Incubate at 37 °C for 1 h in a thermocycler, then cool it down in a ramp from 95 to 25 °C for 5 min (5 °C/min).
Notes:- Alternatively, you can heat up the oligonucleotides and let them cool down at room temperature for several hours.
- Normally phosphorylation is not necessary; however it is required if the plasmid is dephosphorylated.
- You can check if the oligonucleotides are annealed through running a 2% agarose gel using the individual oligonucleotides as controls. Normally we skip this step and have no issues with the ligation afterward.
- Store the annealed oligonucleotides at -20 °C or proceed to the ligation step.
- Design and order oligonucleotides as separate strands (Our oligonucleotides are ordered from Sigma-Aldrich) (Table 1).
- p.UTA.2.0 plasmid Linearization
- Prepare the digestion in a 1.5 ml microfuge tube (Table 3). Scale up accordingly for multiple reactions.
Table 3. Digestion of the plasmid p.UTA.2.0 Empty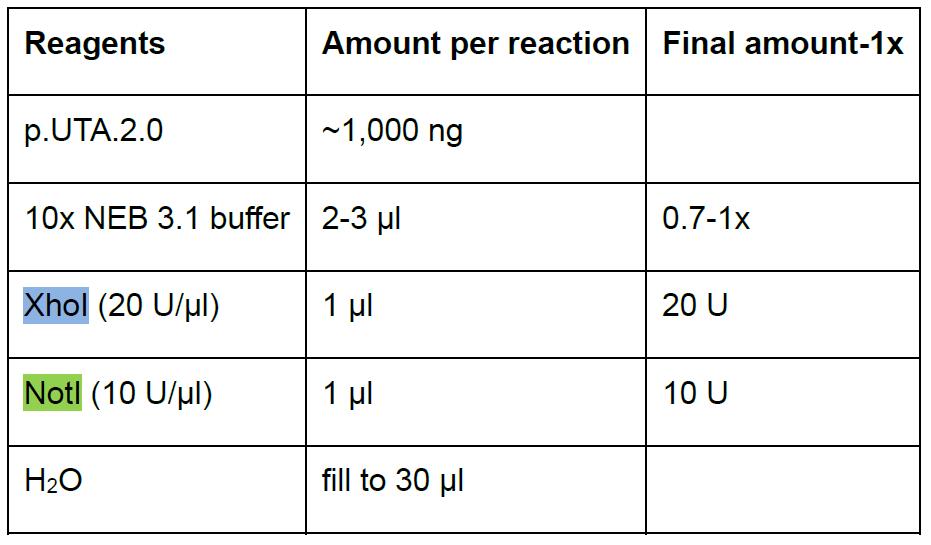
Although not required, we recommend making NotI and XhoI separated controls.
Usually dephosphorylation is not necessary; however, it reduces the background after ligation (see below), in that case, the annealed oligonucleotides need to be phosphorylated. - Vortex briefly to mix.
- Spin the tube quickly to collect the liquid at the bottom.
- Incubate the mixture at 37 °C for at least 1 h.
Note: Overnight incubation is possible. - Run the reactions in a 1% agarose gel.
- Cut the 5,131 bp band and transfer the piece to a clean microcentrifuge tube.
- Extract the DNA from the gel using the QIAquick Gel Extraction Kit following the manufacturer’s protocol.
- Measure DNA concentration.
- Prepare 50 ng/μl aliquots of the linearized plasmid.
- Prepare the digestion in a 1.5 ml microfuge tube (Table 3). Scale up accordingly for multiple reactions.
- Ligation of p.UTA.2.0 and annealed oligo-duplex
- Prepare ligation in a 1.5 ml microfuge tube according to Table 4. Scale up accordingly for multiple reactions.
Table 4. Ligation reaction p.UTA.2.0 and annealed oligos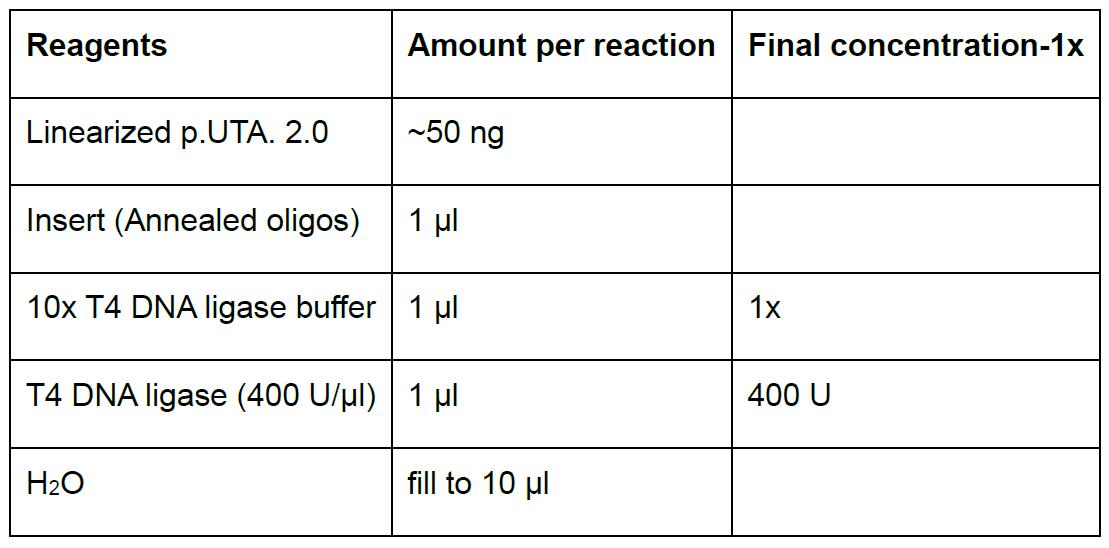
- Vortex briefly.
- Spin the tube quickly to collect the liquid at the bottom of the tube.
- Incubate the mixture at 16 °C for at least 2 h.
Note: Overnight incubation is possible at 4 °C. - Transform into your favorite competent cells.
- Prepare ligation in a 1.5 ml microfuge tube according to Table 4. Scale up accordingly for multiple reactions.
- Screening of positive clones
Notes:- To check for the correct oligonucleotide insertion, we performed endonuclease restriction to distinguish empty clones (w/o miRNA response element) from the positive ones. Since the insertion site (XhoI and NotI) contains an EcoRI site that disappears after digestion, this holds true if the oligonucleotides lack EcoRI sequence.
- We used HindIII and EcoRI together: the empty vector yields four bands (3,992; 1,010; 732; 111) and the positive clones three (3,292; 1,736; 111).
- Since our plasmid lacks a stuffer sequence, we can differentiate background from partially digested DNA; for that reason, we highly recommend performing this screening test (Figure 3).
- Sequences can be retrieved from Addgene Page (http://www.addgene.org/82446/).
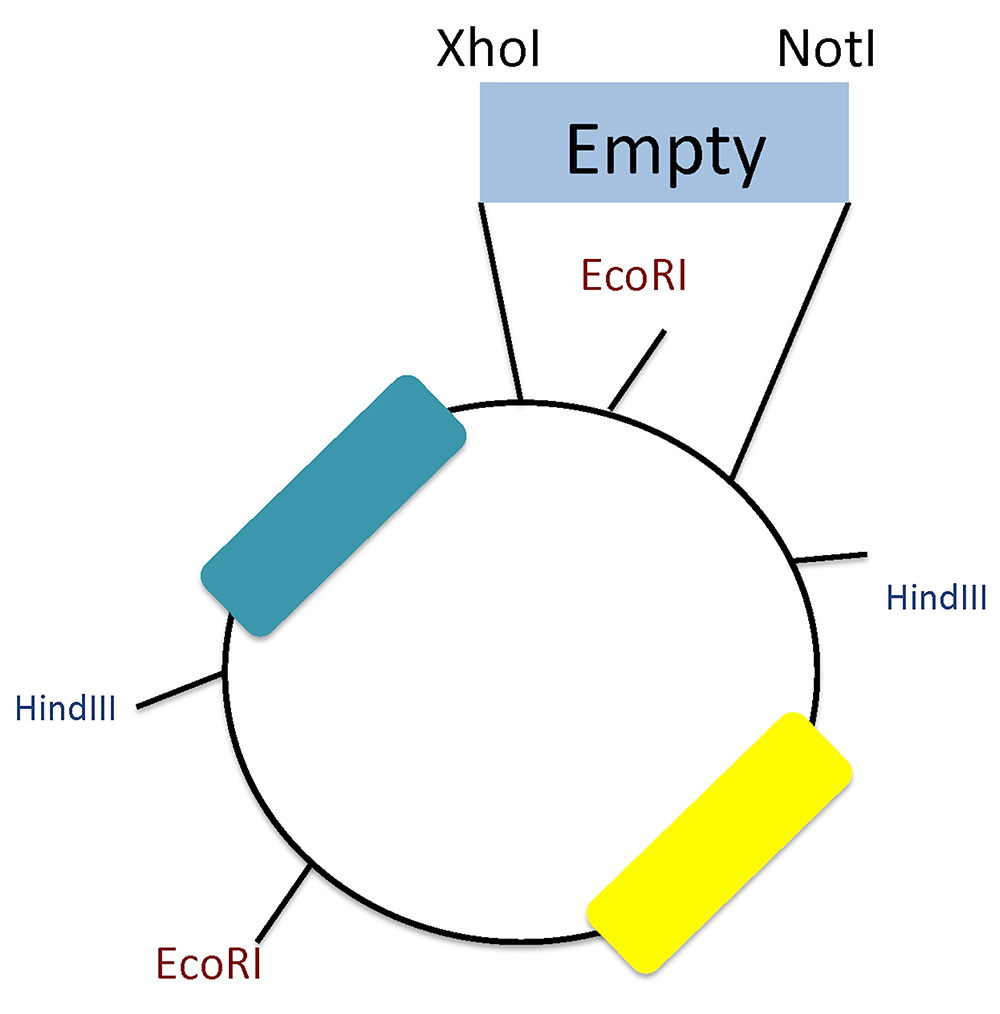
Figure 3. Plasmid scheme and restriction sites. Screening of positive clones using HindIII and EcoRI restriction sites. Notice the EcoRI site between the XhoI and NotI that differentiates between inserted oligos and re-ligation background.
- Grow your bacteria cultures and isolate plasmid using QIAprep Spin Miniprep.
Note: It’s also ok to use home-made solutions to isolate plasmids. - Prepare the digestion reaction in a 1.5 ml microfuge tube (Table 5). Scale up accordingly when performing multiple reactions.
Table 5. Digestion reaction to screen positive clones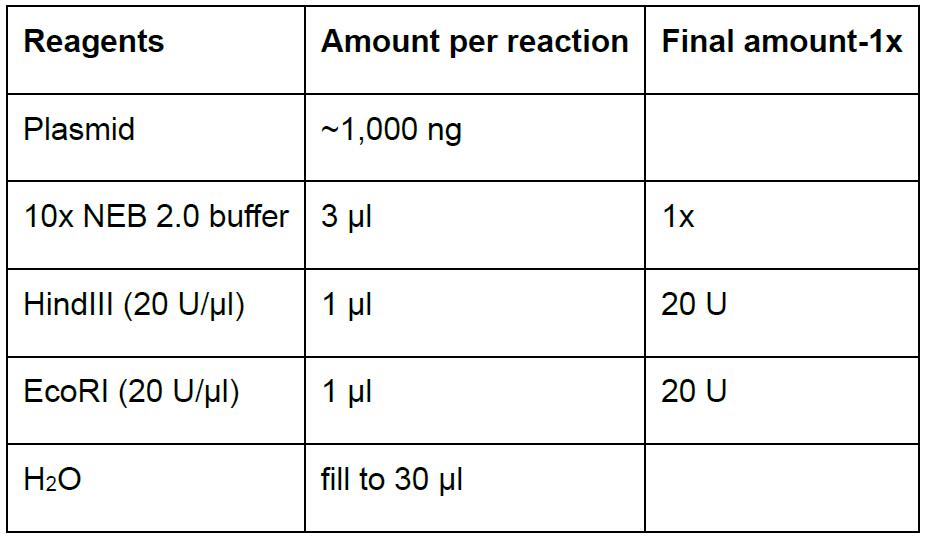
- Vortex briefly and spin to collect liquid at the bottom of the tube.
- Incubate the mixture at 37 °C for 1 h.
- Run the mixtures in a 1% agarose gel.
- Visualize the agarose gel.
- Analyze your clones.
Figure 4 depicts the results of an experiment containing several empty clones derived probably from incomplete digestion of the plasmid. - Prepare transfection quality DNA.
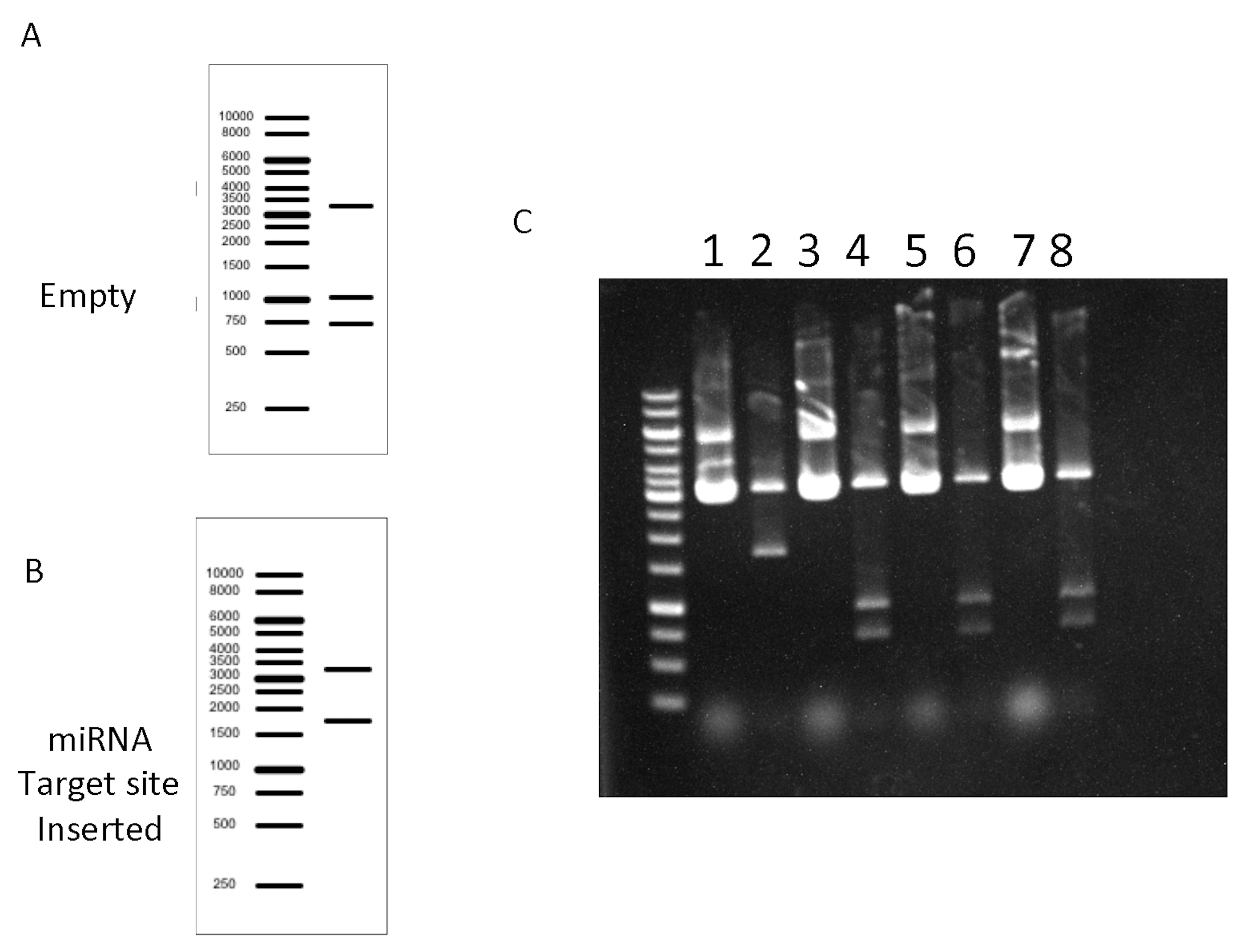
Figure 4. Screening reaction Expected results. A and B. In silico DNA gel electrophoresis of the Empty and the Positive (miRNA target inserted) plasmids. C. Agarose DNA gel electrophoresis from screening reactions, it shows one positive clone (lane 2) and three empty vectors without insert (lanes 4, 6, 8). Non-digested controls (lanes 1, 3, 5, 7).
- Transfection of the p.UTA.2.0 plasmids
This transfection scheme is optimized for 24-well plates using Hek293 cells; we have evaluated this protocol in HeLa, HTC116, and SH-SY5Y. It showed that the neuroblastoma cell line (SH-SY5Y) is not suitable for this assay because the promoters are not strong enough to produce proper analytical functions.- Seed HEK293 cells (about 100,000 cell per well) in a 24-well plate.
- Incubate for 18-24 h at 37 °C and 5% CO2.
- Prepare DNA samples as follows (we recommend at least triplicates per experimental unit. Scale up accordingly):
- Prepare Lipofectamine® 2000 reagent as follows:

- Add the DNA solution (Step E3) to Lipofectamine® 2000 reagent (Step for E4) in a 1:1 ratio.
- Incubate the mixture for at least 20 min at room temperature.
- Add the DNA-lipofectamine mixture dropwise to the cells.
- Incubate for 72 h at 37 °C and 5% CO2.
- Sample preparation for flow cytometry
- Discard medium from the well.
- Wash 1 time with PBS (~500 μl).
- Add 100 μl trypsin per well.
- Incubate for 5 min at 37 °C and 5% CO2.
- Add 400 μl DMEM medium + 15% FCS.
- Transfer the cell suspension to a FACS-tube.
- Centrifuge at 300 x g for 5 min.
- Discard the supernatant.
- Add 1 ml PBS.
- Centrifuge at 300 x g for 5 min.
- Repeat Steps F9 and F10 two more times.
- Discard the supernatant.
- Spin down briefly.
Note: Cells can be documented at this point without fixing. - Add 100 μl PBS.
- Add 100 μl PFA 4% (Paraformaldehyde 4%).
- Incubate for 30 min at 4 °C.
- Wash three times as before (Steps F9 and F10).
- Add 200 μl PBS.
- Seal the FACS-tubes with parafilm.
- Store at 4 °C (fridge).
Note: The fixed cells can be stored up to 1 week without affecting the output of the transfer functions.
Data analysis
- Data Acquisition
The following steps describe the process of gating, and how to export the FCS files to be analyzed using R (It’s assumed that the readers have a basic knowledge of flow cytometry data acquisition). We performed the measurements using a BD LSR II instrument with filters 550LP-BP575/26 (PE) for YFP and BP450/50 (Pacific Blue) for CFP.- Analyze your negative control.
- Set up the single cells using FSC.A and FSC.H scatter plot (Called here P1 or single cells).
- Select the main population of cells using the FSC.A and SSC.A scatter. (Called here P2 or main population).
- Define the YFP positive cells using the histogram visualization (Called here P3 or YFP positive cells). (Figure 5 and Video 1)
Note: Figure 5 and video EmptyFACSdiva (Video 1) show the gates we used for a mock control, and the layout we kept on the acquisition software.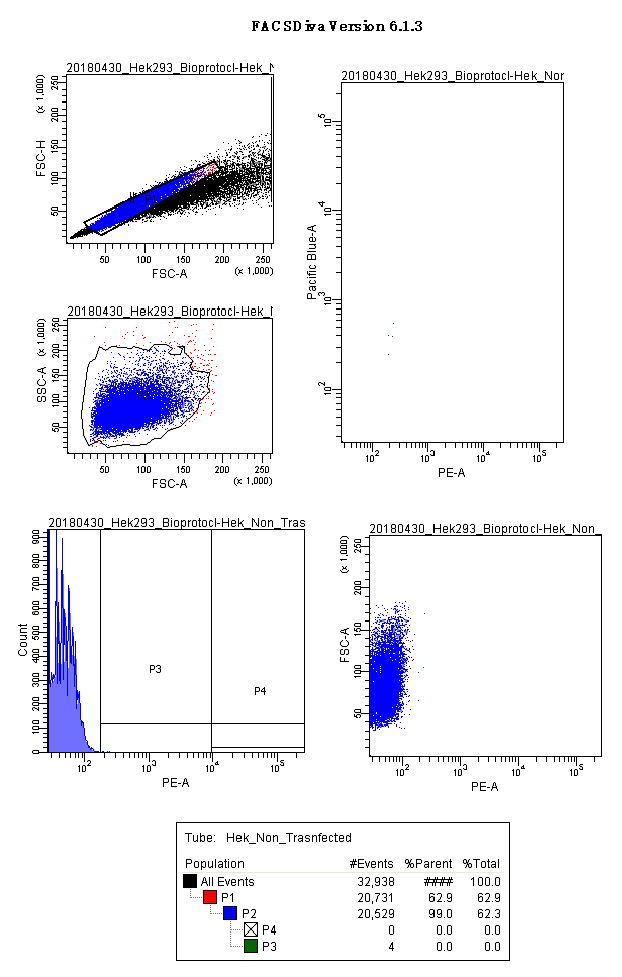
Figure 5. Negative control acquisition (screenshot). Panels show the gates used for HEK293 cells: scatter plots for singlets (FSC.A vs. FSC.H), size and complexity (FSC.A vs. SSC.A); histograms and scatter plot for PE.A (550LP-BP575/26), and the output for CFP and YFP (Scatter plot PE.A vs. Pacific blue). Video 1. FACS Diva acquisition Non-Transfected negative control. Panels show the gates used for HEK293 cells: scatter plots for singlets (FSC.A vs. FSC.H), cell population (FSC.A vs SSC.A), histograms and scatter plot for PE.A (550LP-BP575/26), and the analytical output for CFP and YFP (Scatter plot PE.A vs. Pacific blue).
Video 1. FACS Diva acquisition Non-Transfected negative control. Panels show the gates used for HEK293 cells: scatter plots for singlets (FSC.A vs. FSC.H), cell population (FSC.A vs SSC.A), histograms and scatter plot for PE.A (550LP-BP575/26), and the analytical output for CFP and YFP (Scatter plot PE.A vs. Pacific blue). - Run the p.UTA.2.0 empty vector and create a gate with the Relative Fluorescence Units of PE.A from 10,000 till the end of the x-axis. (Called here P4 or acquisition gate) (Figure 6).
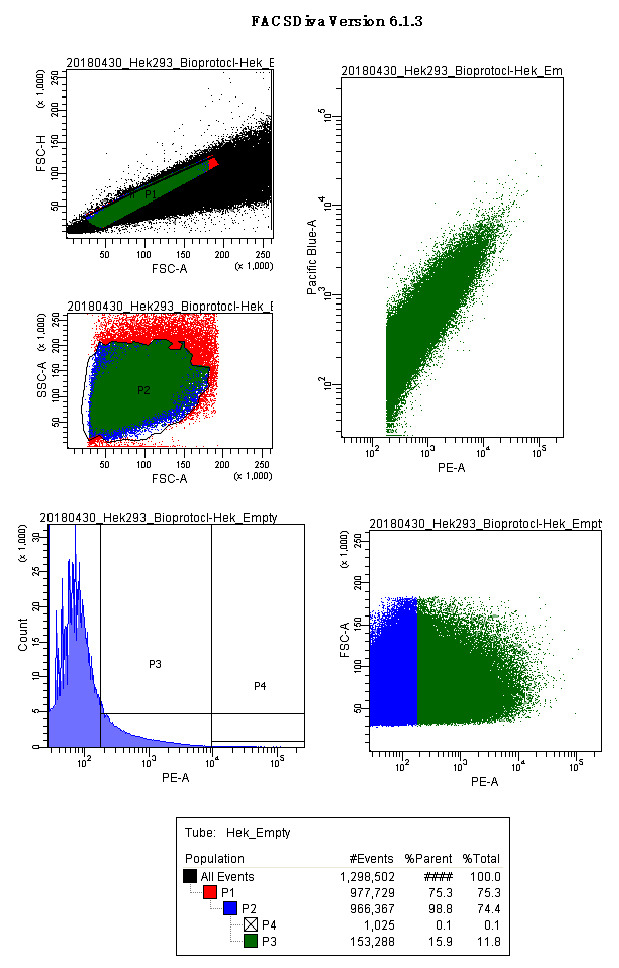
Figure 6. Empty vector control acquisition screenshot. Panels show the gates used for HEK293 cells transfected with the empty vector (p.UTA.2.0 empty). Scatter plots for singlets (FSC.A vs. FSC.H), main cell population (FSC.A vs. SSC.A), histograms and scatter plot for PE.A (550LP-BP575/26), and the analytical output for CFP and YFP (Scatter plot PE.A vs. Pacific blue). - Acquire between 1,000 and 3,000 events from the P4 population.
Note: We used the P4 population as acquisition gate to obtain enough data points at high YFP intensities, reducing the scattering of the analytical function, which essential for the regression analysis. - Read your samples.
Figures 6 and 7 show acquisition panels of HEK293 cells transfected with p.UTA.2.0 Empty and p.UTA.2.0 miR 19b-3p.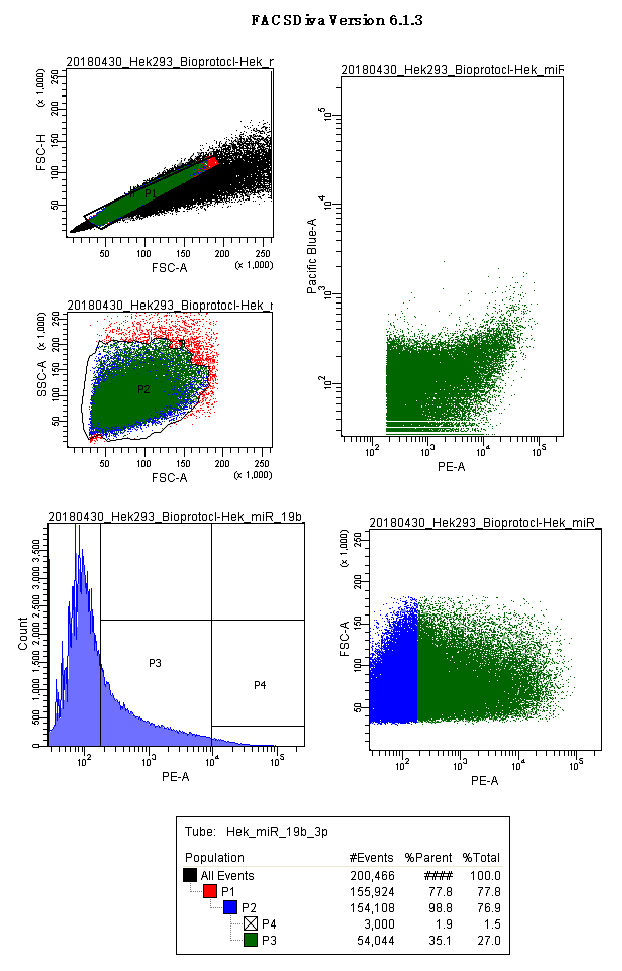
Figure 7. p.UTA.2.0 miR 19b-3p acquisition screenshot. Panels show the gates used for HEK293 cells: scatter plots for singlets (FSC.A vs. FSC.H), size and complexity (FSC.A vs. SSC.A); histograms and scatter plot for PE.A (550LP-BP575/26), and the output for CFP and YFP (Scatter plot PE.A vs. Pacific blue). - Export the P3 gate in FCS format 2.0 (Figure 8).
Note: P3 includes all the YFP positive cells. We used P4 as the stopping gate to improve the quality of the analytical functions. Normally, we also save the whole data set for our records.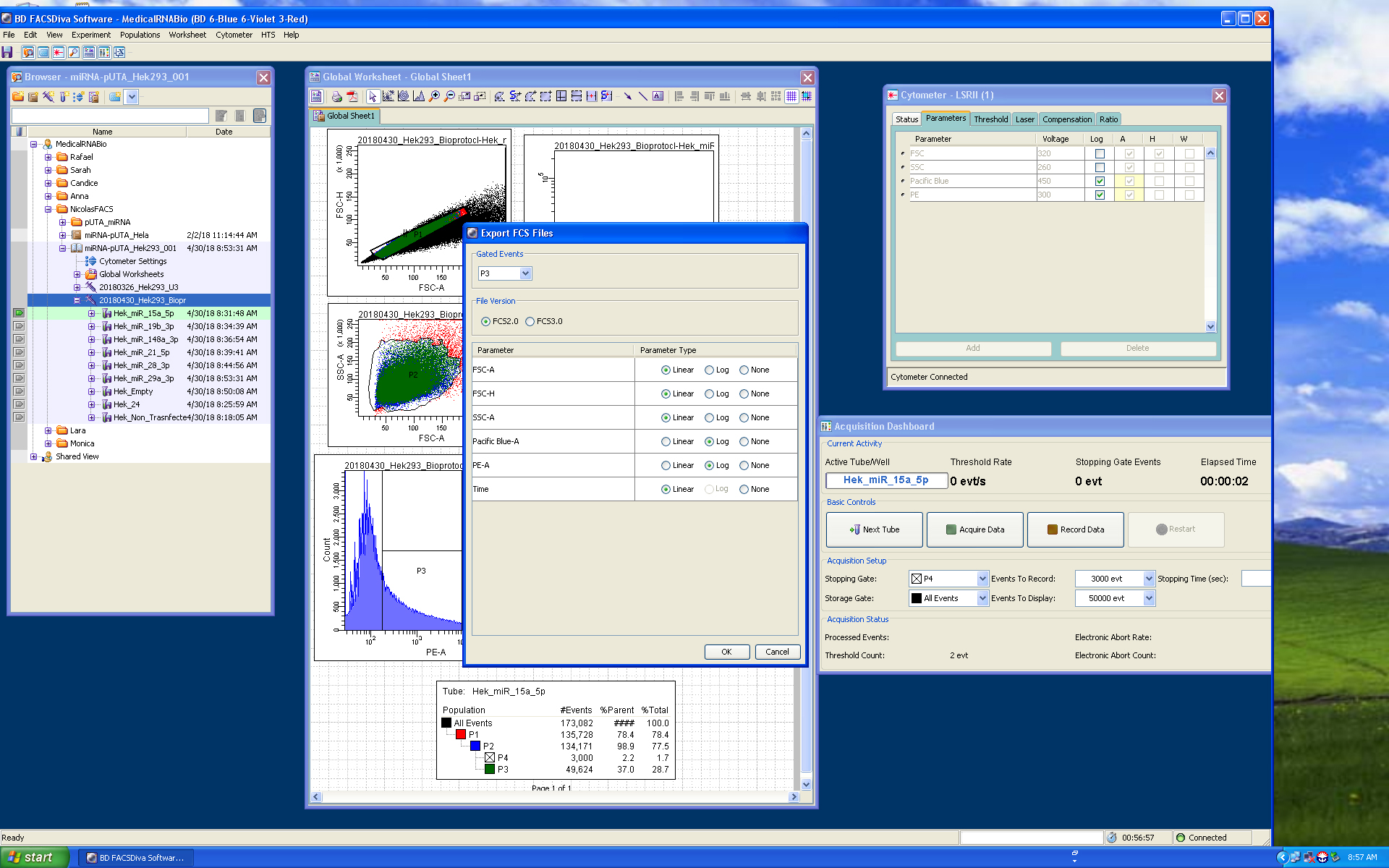
Figure 8. FACS-Diva export data screenshot. Export FCS format 2.0 for P3 (YFP positive cells including P4) population, screenshot exemplifies the export options used for the FCS files.
- Data processing and plotting
Here is a simplified code to efficiently analyze UTA analytic cytometry written for laboratory scientist with minimum or no coding experience. The example FSC files and the codes used in the following steps are included in Data codes.zip and the Video 2 describes each step. Video 2. R studio data analysis
Step by step guide to generate analytical function from Raw FCS files using the R environment:- Locate the FCS files (containing only the YFP positive population) in the same folder with the R script “Analysis_Functions.R” file.
- In R, set your working directory.
setwd("/path/toyour/FCSfiles/Folder”) - Call the functions in the “Analysis_functions_4.0.R” file.
Source("Analysis_functions_4.0.R") - Create a vector containing the FCS files’ names, in this case just files containing YFP positive events, in this example data is the P3 population (Including P4).
fileNames <-Sys.glob("*_P3.fcs") - Create your working List.
The following line will create an R object (List) that contains all the analytical functions for your FCS files. You will apply the bin.data function to each file on your working directory; it means that each object inside the list corresponds to an FCS file or experimental unit. Each FCS processed object will have:- Raw data (CFP, YFP, ratio, fold).
- Bin data (Analytical Function).
- Bin CFP and YFP will be used for Analytical function generation.
- Ratio of CFP/YFP per each CFP bin.
- Fold Repression per each bin. It just another way to see repression; it means that CFP is being repressed at 100%.
- Descriptive statistics with quartiles median, max, and min.
This is a base R loop: it takes each element on fileNames (FCS files) and applies the bin.data.function on them to create a list. The first argument defines the vector (Containing the FCS file labels) and the second includes the bin.data.FscH function where you can modify the bin size. - Write the names of your FCS files, assign them to your list and write them in a text file.
table_names<-data.frame(fileNames)
write.table(table_names,"List_Index.txt")
names(Functions)<-fileNames
The List_Index.txt file contains the names of your FCS files and their indexes inside the list; we recommend having a hard copy to plot the data. - Plot the analytical functions in R (Figure 9).
plot.miR(Functions,fil_num = c(2,4,6,8),control = 1,ylim = c(1,8),l_y = 8,Leg = c("Empty","miR 148a 3p","miR 19b 3p","miR 24 3p","miR 29a 3p"),main="miRNA" ,col.m=c("black","blue","red","green3","purple"),cex = 0.7,mu.cex = 0.3)plot.miR(Functions,fil_num = c(3,5,7),control = 1,ylim = c(1,8),l_y = 8,Leg = c("Empty","miR 15a 5p","miR 21 5p","miR_28_3p"),main= "miRNA",col.m=c("black","blue","red","green3","purple"),cex = 0.7,mu.cex = 0.3 )
The List_Index.txt file contains the names of your FCS files and their indexes inside your list; we recommend having a hard copy of it when you plot the data. Table 6 describes each argument.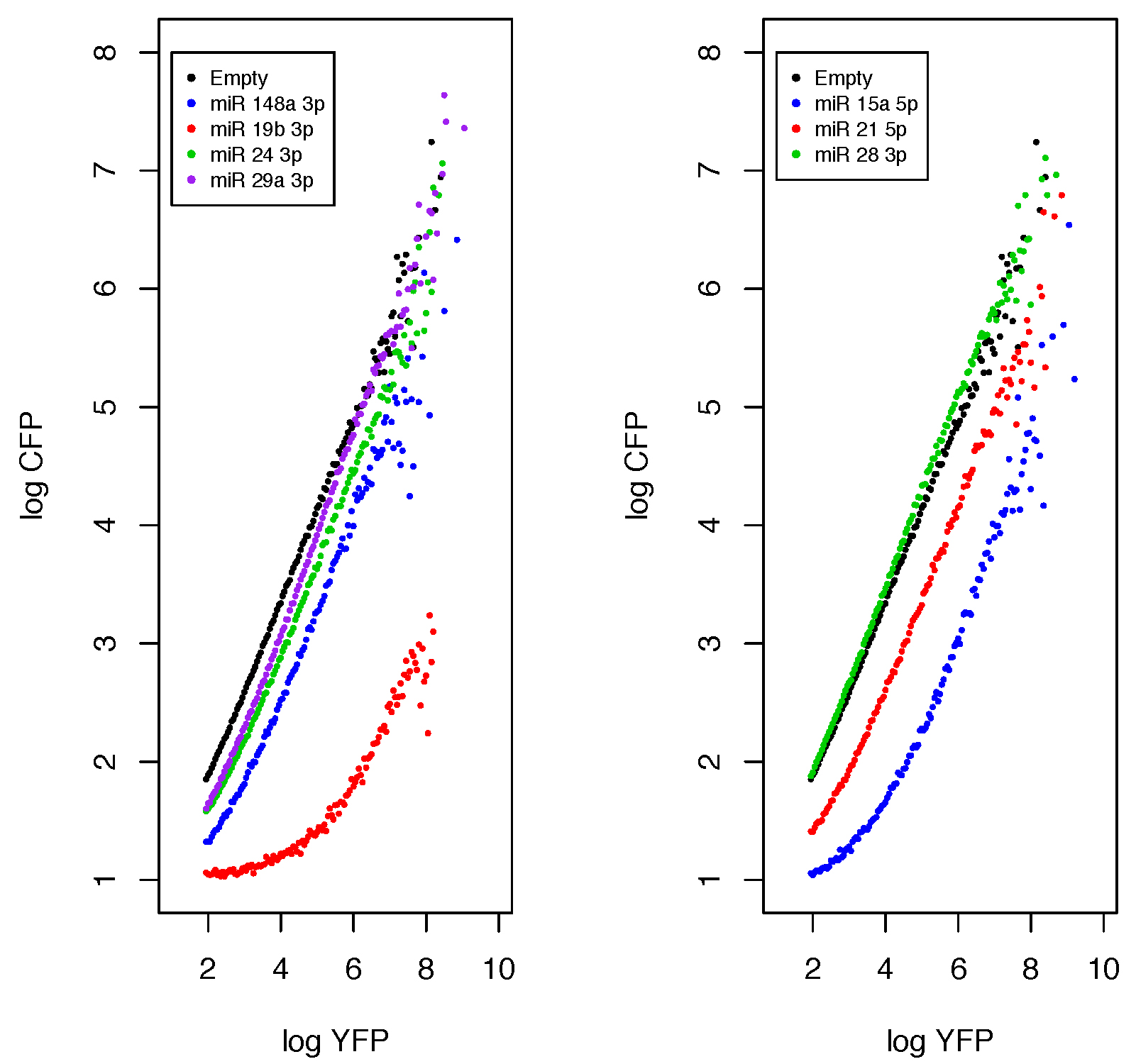
Figure 9. Analytical functions output from the plot.miR function. HEK293 cells were transfected with several miRNA sensors, acquired and read by Flow cytometry. FCS 2.0 files were analyzed in the R environment.
Table 6. The function arguments of plot. miR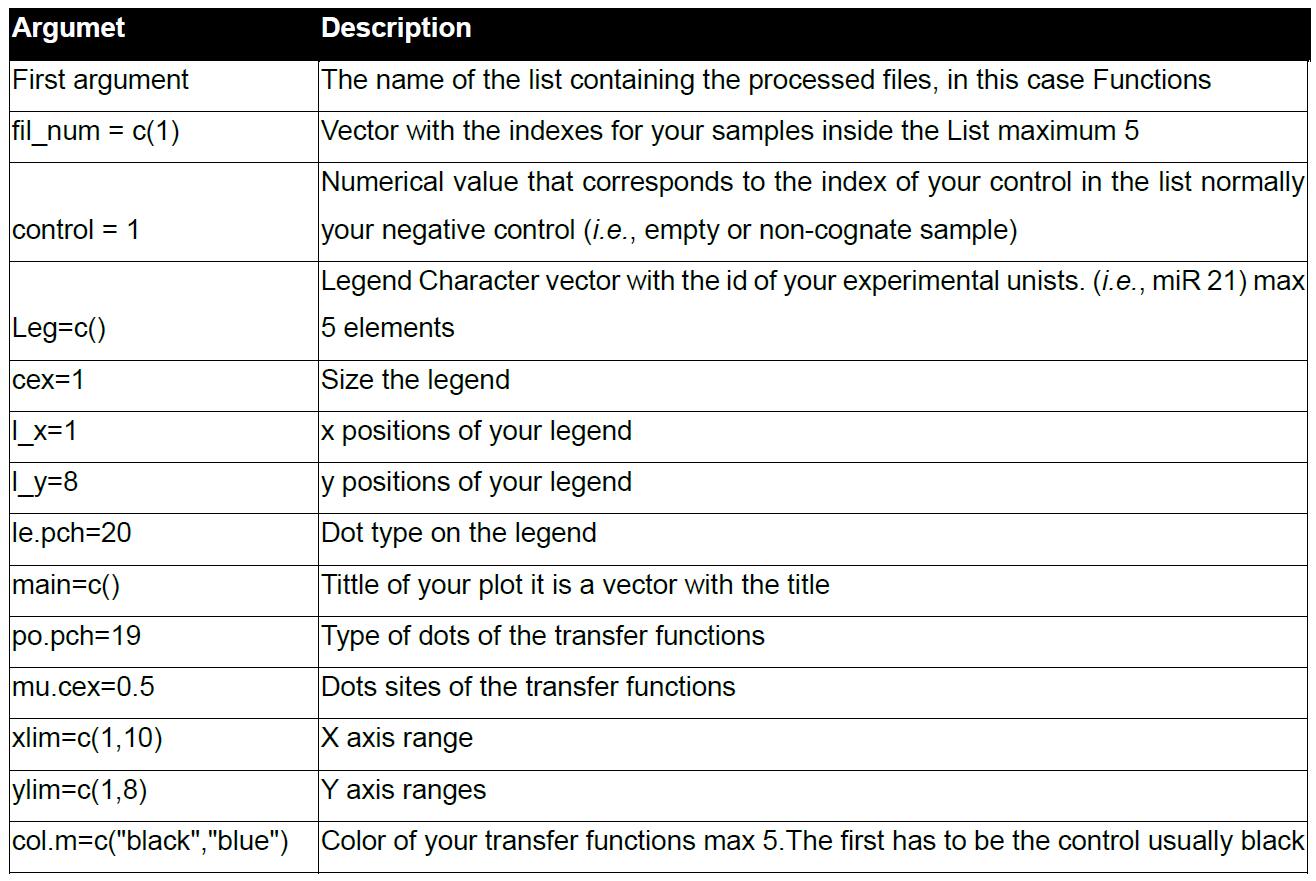
Optional Steps:
You can also generate a PDF with the R plots:- Create the PDF device:
pdf("YourName.pdf",3.18,6.75)
pdf function requires the name of your file in quotes, in this case, “YourName.pdf” do not forget the extension, while the second and third arguments are the width and height of your plot, respectively. - Run your plot codes (more than one is possible, do not expect any output on the R-studio plot layout).plot.miR (Functions, fil_num = c (2,3,4,5),control = 1,ylim = c (1,8),l_y = 8,Leg = c( "Empty","miR_148a_3p","miR_15a_5p","miR_19b_3p","miR_21_5p"),main= "983" ,col.m=c ( "black","blue","red","green3","purple"),cex = 0.7,mu.cex =0.3)
- Close the PDF
dev.off()
You might also use another software to plot your data, using the following function, You will create a CSV file of all your list, the first column has the log values of each YFP bin, and the other columns contain the CFP values of each element in your list (Functions), with the name of your FCS files. - Use the Export function.
Export(Functions,fileNames,"Output.csv")
The Export function requires the list containing the bin data, requires the vector containing the names of your FCS files, which is the same you used to define the index for the plots, the third argument is the name of your CSV file.
- Data interpretation and statistical analysis.
To interpret and gain further insight from the analytical functions, you can extract information using the molecular titration model used before (Mukherji et al., 2011; Lemus-Diaz et al., 2017) (Figure 10).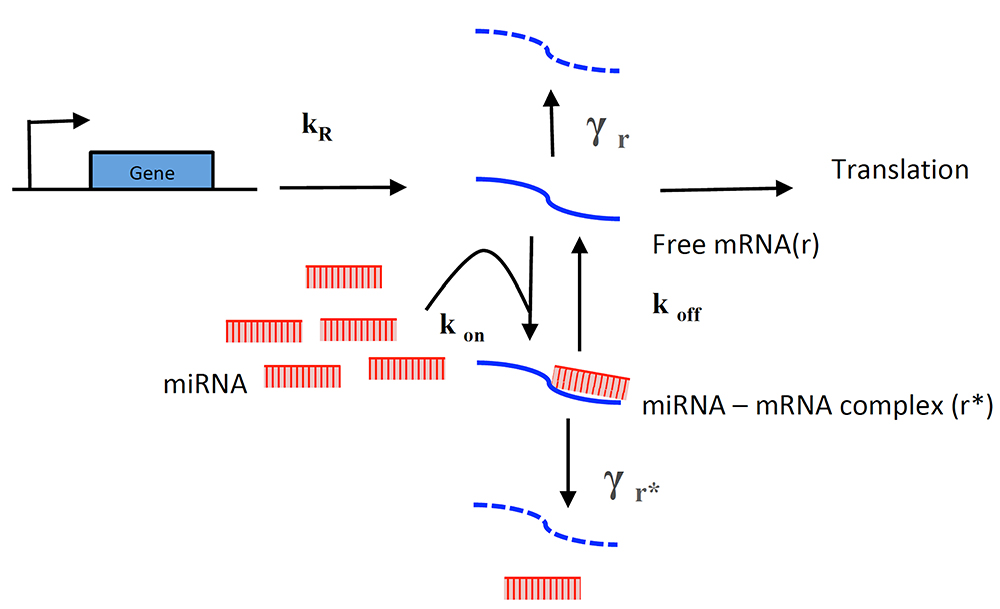
Figure 10. Titration model of miRNA-induced regulation. This model for miRNA-induced regulation describes the free mRNAs available for translation. In the absence of miRNA, the cell transcribes and degrades mRNA at rates KR and γr respectively. In the presence of miRNAs, the free mRNA available for translation depends on the miRNA-mRNA (r*) complex, its formation depends on on-rate (Kon) and miRNA concentration. Then two mutually exclusive paths occur. Either it induces mRNA degradation (at γr* rate) or disassembles into its components (at Koff rate).
The model describes the miRNA titration of mRNA where transcribed mRNA interacts with miRNA (r* complex) ruled by the Kon and Koff of the complex, which depends on the miRNA complementary and target sites in the mRNA. The following equations describe the change of mRNA (free to be translated) and r*:
Equation 1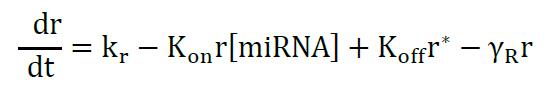
Equation 2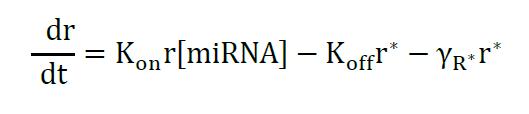
In this context, total miRNA concentration is the free miRNA molecules plus the miRNAs included in the r* complex:
Equation 3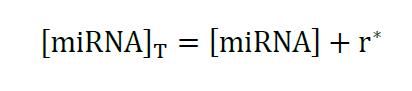
Assuming that no translation happens from miRNA-mRNA complexes, and only from the free RNA (r), the solution for steady state of (r) is given as before (Mukherji et al., 2011; Lemus-Diaz et al., 2017):
Equation 4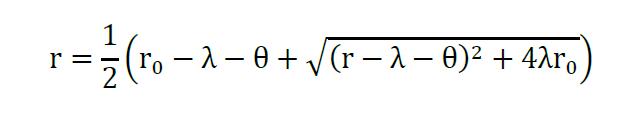
where,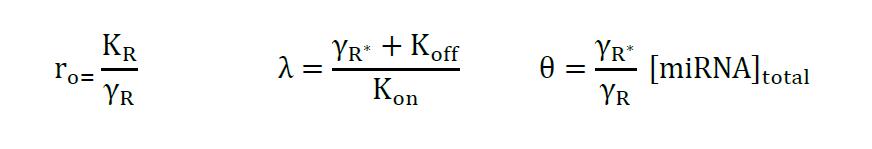
Simulations can be performed using the app (Simulation miRNA function). For instructions, see the supplementary material of Lemus-Diaz et al. (2017).
Using the analytical functions as input (Excel or R) you can estimate the parameters θ and λ, using non-linear regression with equation 4, while θ corresponds miRNA concentration λ correlates with the binding of the miRNA. Estimated parameters for the examples can be found in the supplementary information of Lemus-Diaz et al. (2017).
We validated this model for the reporter described here and showed how the parameters (θ and λ) are useful to dissect miRNA activity, giving as an example how localization of a miRNA could be inferred by the analytical functions (Lemus-Diaz et al., 2017, Figure 6). Moreover, using this model, we described three categorical variables (Low, Mid, and High functional groups) to interpret the outputs easily (Lemus-Diaz et al., 2017, Figure 2).
In the data presented in Figure 9, the miR 24-3p, miR 29a-3p can be classified as low functional; miR 148a-3p and miR 21-5p as mid; and miR 21-5p and miR 19b-3p as highly functional. However, as any qualitative data this classification is arbitrary, so we advise to use the estimate parameters and use them to compare the miRNA functionality (Lemus-Diaz et al., 2017, Figures 3 and 4).
Acknowledgments
This work was partly supported by the Göttingen Graduate School for Neuroscience, Biophysics, and Molecular Biosciences (DFG grant GSC 226/2), L.T was a master student at the IMPRS for Molecular Biology. This protocol was adapted from Mukherji et al. (2011) and Lemus-Diaz et al. (2017); the term Analytical flow cytometry was adopted from Denzler et al. (2016).
Competing interests
The authors declare that there are no conflicts of interest or competing interest.
References
- Bai, B., Liu, H. and Laiho, M. (2014a). Small RNA expression and deep sequencing analyses of the nucleolus reveal the presence of nucleolus-associated microRNAs. FEBS Open Bio 4: 441-449.
- Bai, B., Yegnasubramanian, S., Wheelan, S. J. and Laiho, M. (2014b). RNA-Seq of the nucleolus reveals abundant SNORD44-derived small RNAs. PLoS One 9(9): e107519.
- Bartel, D. P. (2018). Metazoan microRNAs. Cell 173(1): 20-51.
- Bartel, D. P. and Chen, C. Z. (2004). Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 5(5): 396-400.
- Bazzini, A. A., Lee, M. T. and Giraldez, A. J. (2012). Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336(6078): 233-237.
- Bertoli, G., Cava, C. and Castiglioni, I. (2015). microRNAs: New biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics 5(10): 1122-1143.
- Bertoli, G., Cava, C. and Castiglioni, I. (2016). microRNAs as biomarkers for diagnosis, prognosis and theranostics in prostate cancer. Int J Mol Sci 17(3): 421.
- Bosson, A. D., Zamudio, J. R. and Sharp, P. A. (2014). Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 56(3): 347-359.
- Brameier, M., Herwig, A., Reinhardt, R., Walter, L. and Gruber, J. (2011). Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic Acids Res 39(2): 675-686.
- Denzler, R., McGeary, S. E., Title, A. C., Agarwal, V., Bartel, D. P. and Stoffel, M. (2016). Impact of microRNA levels, Target-Site complementarity, and cooperativity on competing endogenous RNA-regulated gene expression. Mol Cell 64(3): 565-579.
- Di Leva, G. and Croce, C. M. (2013). miRNA profiling of cancer. Curr Opin Genet Dev 23(1): 3-11.
- Ender, C., Krek, A., Friedlander, M. R., Beitzinger, M., Weinmann, L., Chen, W., Pfeffer, S., Rajewsky, N. and Meister, G. (2008). A human snoRNA with microRNA-like functions. Mol Cell 32(4): 519-528.
- Fabian, M. R., Sonenberg, N. and Filipowicz, W. (2010). Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 79: 351-379.
- Garcia, D. M., Baek, D., Shin, C., Bell, G. W., Grimson, A. and Bartel, D. P. (2011). Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol 18(10): 1139-1146.
- Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., Ellis, B., Gautier, L., Ge, Y., Gentry, J., Hornik, K., Hothorn, T., Huber, W., Iacus, S., Irizarry, R., Leisch, F., Li, C., Maechler, M., Rossini, A. J., Sawitzki, G., Smith, C., Smyth, G., Tierney, L., Yang, J. Y. and Zhang, J. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5(10): R80.
- Grosswendt, S., Filipchyk, A., Manzano, M., Klironomos, F., Schilling, M., Herzog, M., Gottwein, E. and Rajewsky, N. (2014). Unambiguous identification of miRNA: target site interactions by different types of ligation reactions. Mol Cell 54(6): 1042-1054.
- Hafner, M., Landthaler, M., Burger, L., Khorshid, M., Hausser, J., Berninger, P., Rothballer, A., Ascano, M., Jr., Jungkamp, A. C., Munschauer, M., Ulrich, A., Wardle, G. S., Dewell, S., Zavolan, M. and Tuschl, T. (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141(1): 129-141.
- Hahne, F., LeMeur, N., Brinkman, R. R., Ellis, B., Haaland, P., Sarkar, D., Spidlen, J., Strain, E. and Gentleman, R. (2009). flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics 10: 106.
- Hasler, D., Lehmann, G., Murakawa, Y., Klironomos, F., Jakob, L., Grasser, F. A., Rajewsky, N., Landthaler, M. and Meister, G. (2016). The lupus autoantigen la prevents mis-channeling of tRNA fragments into the human microRNA pathway. Mol Cell 63(1): 110-124.
- Helwak, A., Kudla, G., Dudnakova, T. and Tollervey, D. (2013). Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153(3): 654-665.
- Huber, W., Carey, V. J., Gentleman, R., Anders, S., Carlson, M., Carvalho, B. S., Bravo, H. C., Davis, S., Gatto, L., Girke, T., Gottardo, R., Hahne, F., Hansen, K. D., Irizarry, R. A., Lawrence, M., Love, M. I., MacDonald, J., Obenchain, V., Oles, A. K., Pages, H., Reyes, A., Shannon, P., Smyth, G. K., Tenenbaum, D., Waldron, L. and Morgan, M. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12(2): 115-121.
- Krishnan, P., Ghosh, S., Wang, B., Li, D., Narasimhan, A., Berendt, R., Graham, K., Mackey, J. R., Kovalchuk, O. and Damaraju, S. (2015). Next generation sequencing profiling identifies miR-574-3p and miR-660-5p as potential novel prognostic markers for breast cancer. BMC Genomics 16: 735.
- Lagos-Quintana, M., Rauhut, R., Lendeckel, W. and Tuschl, T. (2001). Identification of novel genes coding for small expressed RNAs. Science 294(5543): 853-858.
- Lai, X., Wolkenhauer, O. and Vera, J. (2016). Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res 44(13): 6019-6035.
- Lemus-Diaz, N., Boker, K. O., Rodriguez-Polo, I., Mitter, M., Preis, J., Arlt, M. and Gruber, J. (2017). Dissecting miRNA gene repression on single cell level with an advanced fluorescent reporter system. Sci Rep 7: 45197.
- Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B. L., Mak, R. H., Ferrando, A. A., Downing, J. R., Jacks, T., Horvitz, H. R. and Golub, T. R. (2005). MicroRNA expression profiles classify human cancers. Nature 435(7043): 834-838.
- Moore, M. J., Scheel, T. K., Luna, J. M., Park, C. Y., Fak, J. J., Nishiuchi, E., Rice, C. M. and Darnell, R. B. (2015). miRNA-target chimeras reveal miRNA 3'-end pairing as a major determinant of Argonaute target specificity. Nat Commun 6: 8864.
- Mukherji, S., Ebert, M. S., Zheng, G. X., Tsang, J. S., Sharp, P. A. and van Oudenaarden, A. (2011). MicroRNAs can generate thresholds in target gene expression. Nat Genet 43(9): 854-859.
- Muller, S., Raulefs, S., Bruns, P., Afonso-Grunz, F., Plotner, A., Thermann, R., Jager, C., Schlitter, A. M., Kong, B., Regel, I., Roth, W. K., Rotter, B., Hoffmeier, K., Kahl, G., Koch, I., Theis, F. J., Kleeff, J., Winter, P. and Michalski, C. W. (2015). Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol Cancer 14: 94.
- Salomon, W. E., Jolly, S. M., Moore, M. J., Zamore, P. D. and Serebrov, V. (2015). Single-molecule imaging reveals that Argonaute reshapes the binding properties of its nucleic acid guides. Cell 162(1): 84-95.
- Schramedei, K., Morbt, N., Pfeifer, G., Lauter, J., Rosolowski, M., Tomm, J. M., von Bergen, M., Horn, F. and Brocke-Heidrich, K. (2011). MicroRNA-21 targets tumor suppressor genes ANP32A and SMARCA4. Oncogene 30(26): 2975-2985.
- Steinkraus, B. R., Toegel, M. and Fulga, T. A. (2016). Tiny giants of gene regulation: experimental strategies for microRNA functional studies. Wiley Interdiscip Rev Dev Biol 5(3): 311-362.
- Vera, J., Lai, X., Schmitz, U. and Wolkenhauer, O. (2013). MicroRNA-regulated networks: the perfect storm for classical molecular biology, the ideal scenario for systems biology. Adv Exp Med Biol 774: 55-76.
- Wee, L. M., Flores-Jasso, C. F., Salomon, W. E. and Zamore, P. D. (2012). Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell 151(5): 1055-1067.
- Yang, Y., Chaerkady, R., Beer, M. A., Mendell, J. T. and Pandey, A. (2009). Identification of miR-21 targets in breast cancer cells using a quantitative proteomic approach. Proteomics 9(5): 1374-1384.
- Yu, F., Bracken, C. P., Pillman, K. A., Lawrence, D. M., Goodall, G. J., Callen, D. F. and Neilsen, P. M. (2015). p53 represses the oncogenic sno-miR-28 derived from a snoRNA. PLoS One 10(6): e0129190.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lemus-Diaz, N., Tamon, L. and Gruber, J. (2018). Dual Fluorescence Reporter Based Analytical Flow Cytometry for miRNA Induced Regulation in Mammalian Cells. Bio-protocol 8(17): e3000. DOI: 10.21769/BioProtoc.3000.
Category
Molecular Biology > RNA > RNA interference
Cell Biology > Cell-based analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.