- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

3D Culture Protocol for Testing Gene Knockdown Efficiency and Cell Line Derivation

Published: Vol 8, Iss 11, Jun 5, 2018 DOI: 10.21769/BioProtoc.2874 Views: 10862
Reviewed by: Chiara AmbrogioMauro Sbroggio'Enrico Patrucco
Original research article
The authors used this protocol in:
Apr 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Traditional 2D cell cultures with cells grown as monolayers on solid surface still represent the standard method in cancer research for drug testing. Cells grown in 2D cultures, however, lack relevant cell-matrix and cell-cell interactions and ignore the true three-dimensional anatomy of solid tumors. Cells cultured in 2D can also undergo cytoskeletal rearrangements and acquire artificial polarity associated with aberrant gene expression (Edmondson et al., 2014). 3D culture systems that better mimic the in vivo situation have been developed recently. 3D in vitro cancer models (tumorspheres) for studying cancer stem cells have gained increased popularity in the field (Weiswald et al., 2015). Systems that use matrix-embedded or encapsulated spheroids, spheroids cultured in hanging drops, magnetic levitation systems or 3D printing methods are already being widely used in research and for novel drug screening. In this article, we describe a detailed protocol for testing the effect of shRNA-mediated gene silencing on tumorsphere formation and growth. This approach allows researchers to test the impact of gene knockdown on the growth of tumor initiating cells. As verified by our lab, the protocol can be also used for isolation of 3D cancer cell lines directly from tumor tissues.
Keywords: 3D cultureBackground
3D in vitro cancer cell models represent a bridge experimental method between cell lines and tumors grown in vivo (Pampaloni et al., 2007; Weiswald et al., 2015). 3D characters of solid tumors with heterogeneous access to nutrients or oxygen can only be effectively mimicked by 3D culture systems. In recent years, protocols for tumorsphere culture gained lot of interest. A tumorsphere can be described as a solid, spherical object created from a single progenitor or stem cell. For tumorspheres formation assays, cells are seeded and grown in serum-free media in ultra-low attachment plates (non-adherent conditions), which allows enrichment of cancer cells with stem/progenitor properties (Johnson et al., 2013). Tumorspheres generated from freshly isolated tumor tissue are of special interest in the field because cells from established cell lines typically differ from the primary tumor due to mutations and abnormalities gained during multiple rounds of in vitro passaging. Hereby, we present an optimized protocol for 3D culture-based primary tumor cell isolation and the use of 3D culture to assess the effect of gene silencing on the growth of tumor-initiation cells.
Materials and Reagents
- Eppendorf tube
- Pipette tips
- Vials
- Plastic bottles
- 50 ml Falcon tube
- Petri dishes with clear lid (Fisher Scientific, Fisherbrand, catalog number: FB0875712 )
- 6-well plates, Corning Costar Ultra-Low Attachment (Corning, catalog number: 3471 )
- 15 ml Falcon tubes (Corning, Falcon®, catalog number: 352196 )
- 70 µm cell filter (cell strainer - Corning, Falcon®, catalog number: 352350 )
- Ice
- Plastic pipette
- 24-well plates, Corning Costar Ultra-Low Attachment (Corning, catalog number: 3473 )
- 6-well plates (Corning, catalog number: 3506 )
- Serological pipettes
- Surgical razor blades (FisherBrand High Precision # 22 Style Scalpel Blade, Fisher Scientific, FisherbrandTM, catalog number: 12-000-161 )
- Sterile pipets (10 ml) (FisherBrand Sterile Disposable Standard Serological Pipets, Fisher Scientific, FisherbrandTM, catalog number: 13-678-14A )
- Cryovials (General Long-Term Storage Cryogenic Tubes 1 ml, Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 5000-1012 )
- (optional) Mission pLKO.1-puro-CMV-TurboGFP Positive control Transduction Particles (Sigma-Aldrich, catalog number: SHC003V )
- EDTA (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15575-020 )
- PBS pH 7.4 (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023 )
- Trypsin EDTA (0.05%) (Thermo Fisher Scientific, GibcoTM, catalog number: 25300054 )
- SureEntry transduction reagent (QIAGEN, catalog number: 336921 )
- Clorox Bleach (Veritiv, catalog number: 30966 )
- Heparin Sodium Salt (Sigma-Aldrich, catalog number: H3149-10KU )
- DMEM/F12, Glutamax (Thermo Fisher Scientific, GibcoTM, catalog number: 10565018 )
- Penicillin/Streptomycin (Thermo Fisher Scientific, GibcoTM, catalog number: 15070063 )
- Fetal bovine serum (Thermo Fisher Scientific, GibcoTM, catalog number: 26140079 )
- B27 supplement 50x (serum free) (Thermo Fisher Scientific, GibcoTM, catalog number: 17504044 )
- bFGF – human Fibroblasts Growth Factor 147 basic (animal free) (Gemini Bio-Products, catalog number: 300-805P )
- EGF– Epidermal Growth Factor (human) (Gemini Bio-Products, catalog number: 300-110P )
- ROCK inhibitor (Y-27632) (STEMCELL Technologies, catalog number: 72304 )
- Heparin solution (STEMCELL Technologies, catalog number: 07980 )
- BD Matrigel Matrix Growth Factor Reduced (BD Biosciences, catalog number: 356230 ) or Cultrex® 3-D Culture MatrixTM Reduced Growth Factor Basement Membrane Extract (Trevigen, catalog number: 3445-001-01 )
Notes:- Aliquot Matrigel or Cultrex Matrix into single-use aliquots in a sterile hood into sterile Eppendorf tubes. We recommend 0.5 ml of Matrigel per Eppendorf tube. When combined with 1.5 ml of ice-cold media, this amount is sufficient for 3D culture using one well in a six-well plate.
- Use chilled pipettes and have Eppendorf tubes on ice during procedure.
- Store at -80 °C. Thaw on ice or in the refrigerator overnight before use.
- Aliquot Matrigel or Cultrex Matrix into single-use aliquots in a sterile hood into sterile Eppendorf tubes. We recommend 0.5 ml of Matrigel per Eppendorf tube. When combined with 1.5 ml of ice-cold media, this amount is sufficient for 3D culture using one well in a six-well plate.
- Hibernation medium (HibernateTM-A, Thermo Fisher Scientific, GibcoTM, catalog number: A1247501 )
- Collagenase IV (Sigma-Aldrich, catalog number: C8051-100MG or similar)
- Trypan Blue (TC-10, Bio-Rad Laboratories, catalog number: 1450013 )
- Antibiotic-Antimycotic (100x) (Thermo Fisher Scientific, GibcoTM, catalog number: 15240096 )
- Dispase (1 U/ml, STEMCELL Technologies, catalog number: 07923 )
- EDTA (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15575020 )
- Accutase (STEMCELL Technologies, catalog number: 07920 )
- DMSO (Sigma-Aldrich, catalog number: D2438-50ML )
- Ethanol (70% solution, Fisher Scientific, Fisher BioReagentsTM, catalog number: BP8201500 )
- Culture medium (see Recipes)
- CSC medium (see Recipes)
Equipment
- Centrifuge (temperature controlled, VWR® benchtop general purpose centrifuge) (VWR, catalog number: 10830-764 )
- Pipettes
- Forceps
- CO2 Cell incubator (BINDER, model: CB 160 )
- Biological Safety Level (BSL-2) laminar flow hood (Esco, EscoTechnologies, USA)
- TC20 Automated cell counter (Bio-Rad)
- Mr. Frosty Freezing Container (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 5100-0001 )
- FormaTM 7000 series Ultra Low Temperature Freezers (Thermo Fisher Scientific, Thermo ScientificTM, model: TFM#902 Series )
- Liquid nitrogen storage tank
- Nikon TE-2000E D-Eclipse Csi confocal microscope running Nikon Elements software (Nikon, model: Eclipse TE2000-E )
Software
- Nikon microscope operating software
- ImageJ or similar image processing software (https://imagej.nih.gov/ij/)
- GraphPad Prism 6.0 software from GraphPad Software, USA
(https://www.graphpad.com/scientific-software/prism/) - R software (R Core Team, 2015 R: A language and environment for statistical computing)
Procedure
To better mimic in vivo growth of tumor cells, we used a 3D culture system with extracellular matrix protein mixture for tumorspheres development from a single cell. For testing the effect of shRNA-mediated knockdown of a gene of interest on the growth of tumor cells in 3D culture, prepare cells transduced with virus encoding shControl RNA as well as an shRNA specific for a gene of interest.
Note: In the text, the words ‘spheroids’ and ‘tumorspheres’ are used as synonyms.
- Transduction of cells with virus encoding shRNA
- At the time of transduction, the confluence of the cells should be about 60-70%. Aspirate the medium, wash the plate with sterile PBS and detach cells with Trypsin. Stop the reaction with media containing FBS, centrifuge (300 x g, room temperature; 2 min) and wash the cell pellet with PBS containing SureEntry reagent. For one transduction, use no more than 50,000 cells. Transduce cancer cells in a sterile Eppendorf tube by direct mixing of virus stock solution with cell pellet, washed previously with sterile PBS (pH 7.4) with SureEntry reagent (the final concentration of SureEntry reagent in PBS should be 8 µg/ml). For transduction, use 10 µl of high titer virus (109 TU/ml) per 50,000 cells.
Notes:- Before mixing the pellet with virus solution, aspirate the supernatant leaving small amount (~10 µl) of PBS with SureEntry just above the cell pellet. Discard pipettes and vials containing residual amount of virus into a plastic bottle containing 10% Clorox bleach solution.
- To monitor the efficiency of transduction, control virus can be used (e.g., Mission pLKO.1-puro-CMV-TurboGFP Positive control Transduction Particle, High Titer).
- Transduction of cells is always performed in a Biological Safety Level (BSL-2) laminar flow hood. Use precautions for bloodborne human pathogens and decontaminate the area with 10% bleach.
- Before mixing the pellet with virus solution, aspirate the supernatant leaving small amount (~10 µl) of PBS with SureEntry just above the cell pellet. Discard pipettes and vials containing residual amount of virus into a plastic bottle containing 10% Clorox bleach solution.
- Using forceps carefully transfer the open Eppendorf tube containing the transduction mixture (cells and virus) into a 50 ml Falcon tube. Loosen the cap of Falcon tube to allow gas exchange during incubation. Transfer the Falcon tube containing the Eppendorf tube into an incubator (37 °C, 5% CO2) and incubate for 30 min.
- Remove cells from the incubator and transfer them to a sterile hood. Add sufficient amount of culture media (2 or 3 ml) and plate the cells into a 6-well plate or 10 cm Petri dishes.
- Change the medium next day (discard the aspirated medium into a bottle containing 10% bleach). Allow transduced cells to recover for 2-4 days (the time of recovery depends on confluency of cells).
- Add selection antibiotic at a recommended or pre-tested concentration to get rid of non-transduced cells.
- Keep selection antibiotic in culture medium for 12-14 days.
- Test the efficiency of gene knockown in transduced cells with appropriate methods – qPCR (mRNA level) or Western blot (protein level).
- At the time of transduction, the confluence of the cells should be about 60-70%. Aspirate the medium, wash the plate with sterile PBS and detach cells with Trypsin. Stop the reaction with media containing FBS, centrifuge (300 x g, room temperature; 2 min) and wash the cell pellet with PBS containing SureEntry reagent. For one transduction, use no more than 50,000 cells. Transduce cancer cells in a sterile Eppendorf tube by direct mixing of virus stock solution with cell pellet, washed previously with sterile PBS (pH 7.4) with SureEntry reagent (the final concentration of SureEntry reagent in PBS should be 8 µg/ml). For transduction, use 10 µl of high titer virus (109 TU/ml) per 50,000 cells.
- 3D spheroid culture with cells embedded in ECM protein matrix (Matrigel)
Note: Although more difficult and time consuming, 3D culture with cells/spheroids embedded in matrix is (in our lab) a preferred method for testing the effect of gene silencing on tumorsphere growth. Growing spheroids embedded in solid or semi-solid protein gel matrix do not have the tendency (or the chance) to clump (Figure 1), which makes quantitative analyses (e.g., counting the number of spheroids or measurement of spheroid diameter) difficult.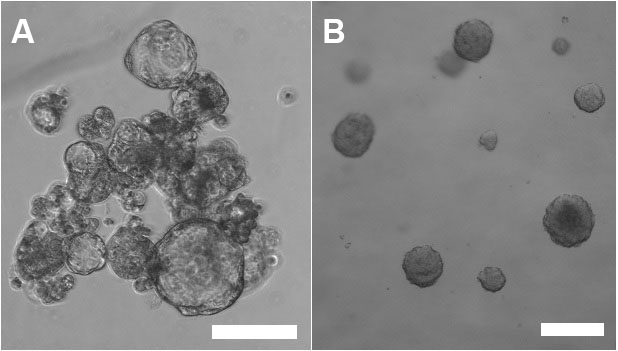
Figure 1. Tumorspheres cultured in CSC media only (left panel, A) or embedded in Matrigel matrix (right panel, B). Note the tendency of tumorspheres to clump when grown in CSC media only. Scale bars represent 50 µm.- Detach cells from 2D plates using Trypsin or other detachment solution for cells. Stop the reaction with media containing FBS and transfer cells from the plate into a 15 ml Falcon tube. Pellet the cells by centrifugation (2 min, 400 x g) at room temperature.
- Aspirate the supernatant and re-suspend the cell pellet in sterile PBS.
- Filter cell suspension with a 70 µm cell filter and count the number of cells manually or using automated cell counter.
Note: The number of cells plated per well must be empirically determined for every type of cancer cells. We recommend starting with 5,000 cells/well (24-well plate) or 10,000 cells/well (6-well plate) and adjusting the cell number as needed (toward lower number of cells per well). - Spin down the cells by centrifugation (2 min, 400 x g) at room temperature.
- Aspirate the supernatant and re-suspend the cell pellet in sterile culture media (abbreviated here as CSC medium) and place the Falcon tube with cells on ice to chill down.
- Pipet ice-cold CSC culture media up-and-down several times to chill the plastic pipette.
- Pipette ice-cold Growth Factor Reduced Matrigel (BD Biosciences, USA) or Cultrex Reduced Growth Factor Basement Membrane Extract, PathClear (Trevigen, USA) on ice. With chilled pipette, transfer Matrigel into the vial containing ice-cold culture media and cells. Use at least 1:3 ratio (1 part of Matrigel with 3 parts of ice-cold media with cells).
- Mix gently, avoid making bubbles.
- Plate the mixture on an ultra-low attachment surface culture plate (Corning, USA) and transfer to the incubator.
- Next day, gently add CSC culture media to cover solidified matrix:media with cells. Add fresh media every 3-5 days.
Note: Aspirate the old media with pipette, not with the vacuum pump (there is a risk of aspiration of Matrigel with spheroids).
- Detach cells from 2D plates using Trypsin or other detachment solution for cells. Stop the reaction with media containing FBS and transfer cells from the plate into a 15 ml Falcon tube. Pellet the cells by centrifugation (2 min, 400 x g) at room temperature.
- 3D spheroid culture (basic, without ECM protein matrix)
- Pellet cells by centrifugation at 300 x g for 2 min (at room temperature) and aspirate the supernatant.
- Mix cell pellet gently with medium (abbreviated here as CSC medium) and plate cells into an ultra-low attachment plate.
Note: The number of cells plated per well must be empirically determined for every type of cancer cells. We typically prepare several wells (with serially diluted cells) to find the concentration of cells that can be easily imaged without problems associated with higher-than-optimal density of cells (clumping, too dense spheroids, etc.). - We recommend starting with 5,000 cells/well (24-well plate) or 10,000 cells/well (6-well plate) and adjusting the cell number as needed.
- Media replenishment can be performed either by adding extra fresh media (this is preferred in first 2-7 days after cell plating during the time spheres are formed) or by changing media completely. To change the media completely, collect the floating cells from the well and centrifuge at 300 x g, for 2 min. Aspirate the supernatant carefully (cell pellet is sometimes not visible, so leaving small amount of media above the pellet is recommended) and add fresh CSC medium. Mix gently and transfer the cells back to the plate.
- Monitor tumorsphere formation daily. Sometimes, cells or spheroids have the tendency to clump in the center of the plate (this is caused by micro vibration of cell culture incubator). In this case, re-suspend the tumorspheres gently by tapping the plate, or mechanically using serological pipettes (in the sterile hood).
- Pellet cells by centrifugation at 300 x g for 2 min (at room temperature) and aspirate the supernatant.
- Establishing a new primary 3D spheroid culture line from human tumor tissue
Note: Before performing an experiment, make sure to obtain an informed consent letter from each patient which acknowledges the use of residual tumor tissue for research. All procedures that involve research with human tumor tissues must be approved by Institutional Review Board (IRB). All collected tissues must be de-identified.- Transfer the native tumor sample from the hospital in sterile PBS or culture media or (the best option) in Hibernation medium (Hibernate®-A, Gibco, Invitrogen), supplemented with Antibiotic-Antimycotic (Gibco, Invitrogen), on ice.
Note: Tumor samples should be as fresh as possible – the ideal situation is to receive/transfer freshly isolated tumor tissue directly from the surgery room – however, in most cases, the tissue used in experiment is residual (leftover) tissue provided by pathologist. - Wash 2 x or 3 x with sterile PBS supplemented with Antibiotic-Antimycotic (100x) (Gibco Invitrogen). Perform final wash with PBS without antibiotics.
- Cut the tumor sample with a surgical blade (Figure 2) into small pieces (1-3 mm) and transfer them into a Falcon tube with ~1 ml of 0.5-1% solution of Collagenase IV (Sigma-Aldrich). Sometimes, collagenase digestion is ‘too strong’ – it is important to monitor sample digestion (take a small portion of the tissue sample in collagenase every 15 min of digestion at 37 °C and check the viability of cells with Trypan blue and TC20 Automated cell counter (Bio-Rad) or equivalent method).
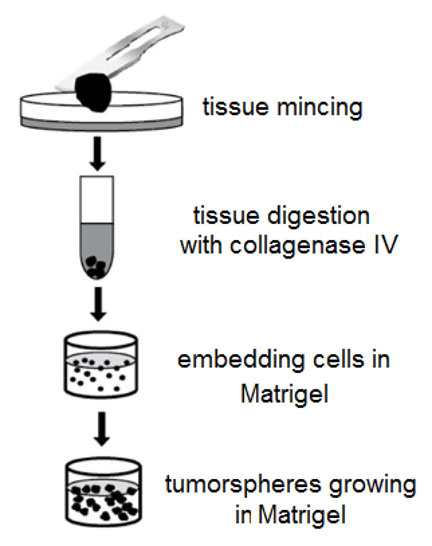
Figure 2. Scheme of tumor tissue processing - Add sterile PBS to the Falcon tube with the tissue sample to wash out the collagenase by centrifugation (200-500 x g, 3 min, room temperature).
- Re-suspend the pellet with sterile PBS and filter cells with 70 µm cell filter. Count cells.
- Re-suspend filtered primary cells with i) CSC media or ii) ice cold CSC media mixed with Matrigel (1:3) and plate into Ultra low attachment plates. Use up to 0.5 x 106 cells per well (for 6-well plate).
- Monitor the growth of spheroids every day. Add small amount of fresh media every 3 days without disturbing the solid phase with embedded cells. Once spheroids are formed, passage them as described below.
Notes:- Sometimes it is difficult to establish 3D culture in serum-free media. We recommend preparing one or two wells with cells cultured in CSC media, supplemented with 1% FBS.
- Freshly digested tumor tissue can be also used for establishing cancer cell lines for 2D growth. In that case, treat the tumor tissue with collagenase as described above. Once digested, wash collagenase with sterile PBS and without filtration the cells, re-suspend tissue fragments/cells in media containing DMEM/F12, 1% Penicillin/Streptomycin , and 10% FBS). Plate into a Petri dish or 6-well plate.
- Sometimes it is difficult to establish 3D culture in serum-free media. We recommend preparing one or two wells with cells cultured in CSC media, supplemented with 1% FBS.
- Change the medium next day to get rid of unattached (dead) cells and debris.
Note: For 2D cell line generation, we do not pass cells through a cell filter – it is better to seed them in clumps and with the ‘trash’ and wash the unattached cells and debris next day when changing the media.
- Transfer the native tumor sample from the hospital in sterile PBS or culture media or (the best option) in Hibernation medium (Hibernate®-A, Gibco, Invitrogen), supplemented with Antibiotic-Antimycotic (Gibco, Invitrogen), on ice.
- Passaging the spheroids
- Aspirate the media from the plate carefully with the pipette, not vacuum aspirator. Try not to disturb the spheroids in Matrigel.
- Collect the Matrigel with embedded spheroids with a 10 ml pipette and transfer it into a fresh 15 ml or 50 ml Falcon Tube (Figure 3).
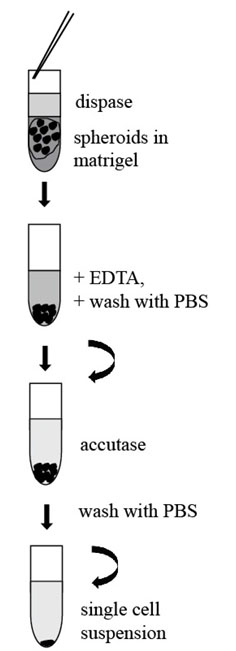
Figure 3. Scheme of procedure for tumorspheres passaging - Add 10 ml of Dispase (1 U/ml, StemCell Technologies, Canada) and place the tube into an incubator (37 °C) for 30-60 min. Tap the tube gently several times during digestion.
- Once spheroids settle at the bottom of the tube, stop the reaction with 100 µl of 0.5 M EDTA (Gibco, Life Technologies, USA) for final EDTA concentration = 0.005 M and centrifuge (1 min, 300 x g, room temperature).
- Discard the supernatant carefully and add 10 ml of sterile PBS to wash. Centrifuge for 1 min (300 x g) and repeat 2 x.
Note: At this point, pelleted tumorspheres can be used for further expansion in Matrigel, isolation of RNA (for qPCR), or preparation of protein lysates for Western blot. - Discard the PBS leaving just a little bit above the pellet containing tumorspheres and add 5 ml of Accutase (StemCell Technologies, Canada). Do not close the Falcon tube tightly to allow gas exchange inside the incubator.
- Place the tube in the incubator (37 °C, 5% CO2) and monitor the enzyme reaction.
Note: To monitor the enzyme reaction, pipette spheroids several times up and down with 10 ml serological pipette. Properly digested spheroids can be easily re-suspended into single cells. - To prepare single cell suspension, re-suspend pellet from digested spheroid with 5 ml serological pipette. Add sterile PBS to wash and centrifuge (300 x g, 2 min, room temperature). Repeat 2 x.
Note: At this point, cells can be used for further expansion in Matrigel, FACS analysis, isolation of RNA (for qPCR), or proteins for Western blot.
- Aspirate the media from the plate carefully with the pipette, not vacuum aspirator. Try not to disturb the spheroids in Matrigel.
- Cryopreservation of tumorspheres
Tumorspheres could be cryopreserved in the form of single cell suspension (prepared with Dispase-Accutase treatment) or tumorspheres (Dispase treatment only).- Mix either single cells (0.5 x 106 to 2 x 106) or tumorspheres (tumorspheres from one well from 6-well plate per one cryovial) with fresh CSC culture media (final volume = 1 ml) containing 10% DMSO (Sigma-Aldrich, USA), and place the cryovials immediately into Mr. Frosty Freezing Container (Thermo Scientific).
- Transfer Mr. Frosty into a -80 °C freezer. Next day, transfer vials with frozen tumorspheres/cells into liquid nitrogen storage tank. Use the vapor phase for long storage of tumorspheres/cells.
- Mix either single cells (0.5 x 106 to 2 x 106) or tumorspheres (tumorspheres from one well from 6-well plate per one cryovial) with fresh CSC culture media (final volume = 1 ml) containing 10% DMSO (Sigma-Aldrich, USA), and place the cryovials immediately into Mr. Frosty Freezing Container (Thermo Scientific).
Data analysis
- Data collection and image analysis
In the original article (Strnadel et al., 2017), the images of growing spheres were obtained with Nikon TE-2000E D-Eclipse Csi confocal microscope running Nikon Elements software. Images of multiple fields were taken from each well (from 5 to 20) from which the diameter and the number of tumorspheres were quantified. The diameter (Figure 4) of every tumorsphere was measured (for calibration of the measurement, the scale bar from Nikon microscope operating software was used as described below.). The diameter of cell clumps and single or dead cells (Figure 5) were not measured and the value for these events was set to be 0 (see Statistical analysis).
Note: Optimally, sufficient number of images to cover the whole well of a culture plate should be taken. If it is not possible, take several representative images from every well. Take the same number of images for shControl and for the shRNA sample.- Take multiple images from 3D culture. Make sure that the image has an embedded scale bar from microscope operating software.
- Open images in ImageJ software. The icon menu bar will pop up.
Note: ImageJ is freely available image processing software (freeware). Similar image processing programs can also be used. - Click on “Oval” selection icon located on ImageJ menu bar.
- Measure the diameter of every sphere (in pixels) by fitting the oval selection gate to each particular spheroid. The width of spheroid represents its diameter (d).
- Measure the length of scale bar (click Straight line icon in ImageJ menu). Compare measured diameter values (in pixels) with a known length of scale bar. Calculate the diameter of every spheroid.

Figure 4. Measurement of tumorspheres diameter. Images were taken and opened in ImageJ program. The width of circle, fitted accurately to a particular tumorsphere represents its diameter (d). Scale bar was used to calibrate the measurement (Scale bar represents 100 µm).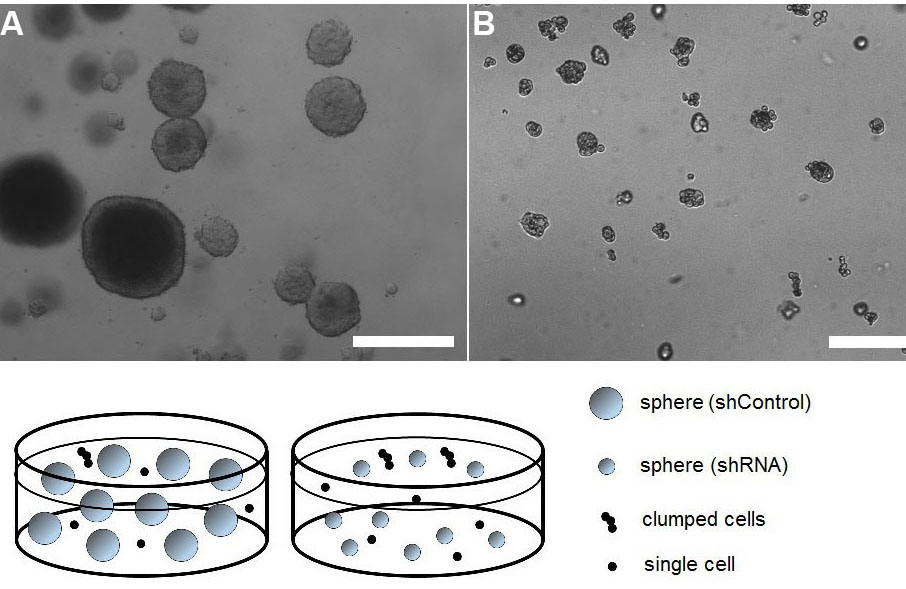
Figure 5. Representative images of tumorspheres established from pancreatic cancer cells transduced with viruses encoding Control shRNA (A) or shRNA of a gene of interest (in this case PEAK1; pseudopodium-enriched atypical kinase 1) (B). The dramatic effect of PEAK1 gene silencing is visible when tumorsphere size is compared between the two groups. Clumped cells and single cell events were excluded from the analysis. Scale bars represent 100 µm.
- Take multiple images from 3D culture. Make sure that the image has an embedded scale bar from microscope operating software.
- Statistical analysis
The data were plotted and analyzed in GraphPad Prism 6.0 software as described in the `Statistical analysees’ section of the original paper (Strnadel et al., 2017). For users more experienced in bio-statistical methods and familiar with R software, we recommend the following strategy of data processing: [Since] the probability density of the sphere diameter is – due to the presence of clumps – bimodal (Figure 6 for an illustration), the Wilcoxon two-sample test seems more appropriate than the t-test used in the original paper. It is also worth mentioning that the equality of the probability of clumps in shControl and shRNA may be tested by the two-sample test for the equality of proportions.
For the illustration, we present results of data analysis for the following data set of tumorsphere diameters.
shControl: 213 µm, 124 µm, 316 µm, 173 µm, 284 µm, 373 µm, 311 µm, 404 µm, 440 µm, 351 µm, 204 µm, 231 µm, 147 µm,151 µm,160 µm, 227 µm, 151 µm, 151 µm, 89 µm, 333 µm, 338 µm, 80 µm, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0 µm).
shRNA: 84 µm, 102 µm, 107 µm, 102 µm, 124 µm, 84 µm, 93 µm, 102 µm, 129 µm, 107 µm, 102 µm, 102 µm, 107 µm, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0 µm).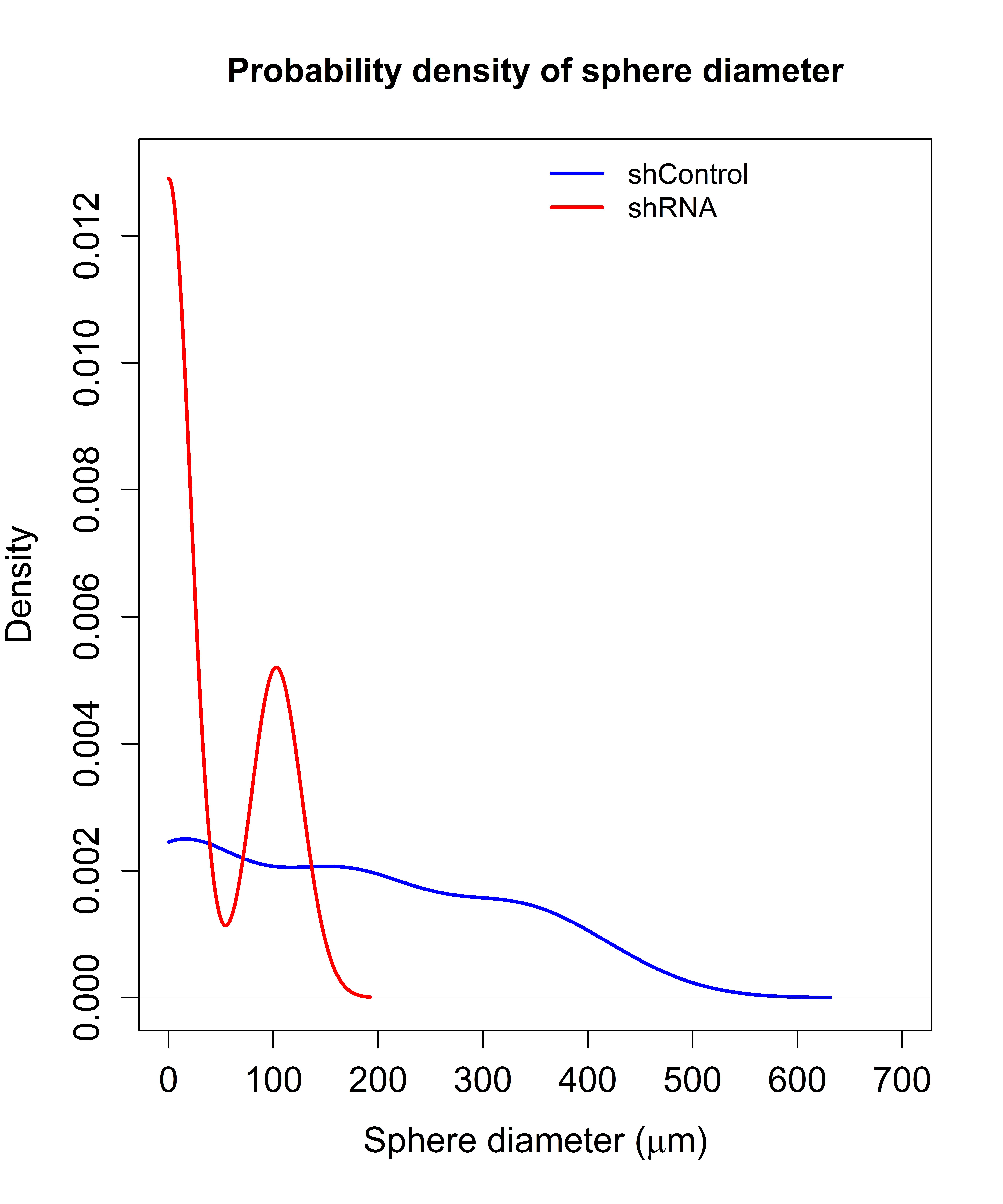
Figure 6. Probability density of sphere diameter, estimated by the kernel density estimator
A kernel density estimate of the probability density of the sphere diameter for shControl and shRNA is shown in Figure 6. A Kernel density estimate is a smoothed histogram. Bi-modality of the distribution, caused by the presence of clumps (i.e., the 2D cells) is clearly visible. Boxplot with swarmplot of the data is presented in Figure 7.
The Wilcoxon test of the equality of the population median of the diameter in shControl and shRNA leads to the P-value of 0.00006 and the difference between the medians of shControl and shRNA is 124.4 µm.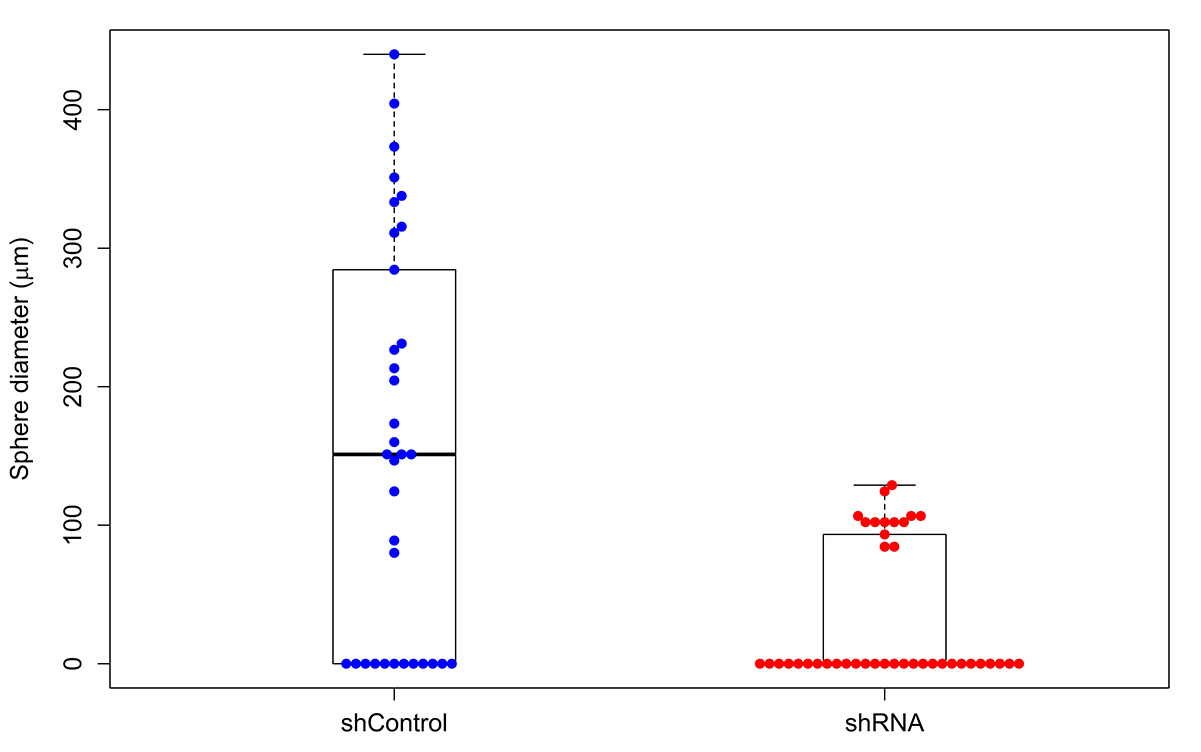
Figure 7. Boxplot with swarmplot of the data
The question whether the population proportion of clumps is the same in shControl as in shRNA, when addressed by the two sample test of proportions, leads to the P-value of 0.1036 and the 95 percent confidence interval for the difference of proportion between shControl and shRNA (-0.5716, -0.0884). The proportion of clumps in shControl is 0.3529, whereas 0.6829 in shRNA population.
The data analysis was performed in R software, using the functions wilcox.test, prop.test, density, boxplot and beeswarm.
Notes
The fresh tumor tissues provided by pathologists are typically non-sterile or contaminated. Based on our experience, we highly recommend sterilizing the tumor tissue before processing. You can sterilize the tissue with 70% Ethanol (submerge the tissue into 70% Ethanol for 3-5 sec or spray the tumor tissue with Ethanol). Wash the tissue with excess amount of sterile PBS immediately after sterilization with Ethanol, and then continue with collagenase treatment.
Recipes
- Culture medium used for transduction of cells with shRNA encoding virus
Note: Mix all of the components in sterile hood and store at 4 °C.
500 ml DMEM/F12, Glutamax (Gibco, Thermo Fisher Scientific, USA)
1% Penicillin/Streptomycin (Gibco, Thermo Fisher Scientific, USA)
10% Fetal bovine serum (Gibco, Thermo Fisher Scientific, USA) - CSC medium
Notes:- Mix all of the components in a sterile hood and store at 4 °C for up to 2 weeks. Add fresh aliquots of bFGF and EGF every time media is used.
- *Store aliquots at -20 °C.
- ** Protect aliquots from light and store at -20 °C.
- Heparin is an anticoagulant (prevents cell clumping) and increases the stability and functionality of FGF. Store at 2-8 °C.
5 ml Penicillin/Streptomycin 100x (Gibco, Life Technologies, USA)
10 ml of B27 supplement 50x (serum free) (Gibco, Life Technologies, USA)
*bFGF – human Fibroblasts Growth Factor 147 basic (animal free) (Gemini Bio-Products, USA), final concentration 20 ng/ml
*EGF– Epidermal Growth Factor (human) (Gemini Bio-Products), final concentration 20 ng/ml
**ROCK inhibitor (Y-27632) (StemCell Technologies, Canada), final concentration 10 µM
Heparin solution (Stemcell Technologies, Canada), final concentration 2 µg/ml - Mix all of the components in a sterile hood and store at 4 °C for up to 2 weeks. Add fresh aliquots of bFGF and EGF every time media is used.
Acknowledgments
These experimental protocols have been previously published and are presented here in original and modified forms from Strnadel et al., 2017. Authors want to thank Prof. Richard Klemke from Department of Pathology (University of California, San Diego) for his support. Special thanks goes to Dr. Vratislav Horak from IAPG, Libechov, Czech Republic, Dr.Michal Kalman, Dr.Juraj Marcinek and Prof.Lukas Plank from Clinic of Surgery and Transplant Center, Jessenius Faculty of Medicine in Martin, and Prof. Ludovit Laca and Dr. Jan Janik from Clinic of Surgery and Transplant Center, Jessenius Faculty of Medicine in Martin, Comenius University in Bratislava.This work was supported by grants, listed in the original article (Strnadel et al., 2017), grants from Research and Development Support Agency (Grant no. APVV‑15‑0217) and the project ‘Biomedical Center Martin’, (ITMS code 26220220187), co‑financed from EU sources. This publication is also the result of the project implementation: "CENTER OF TRANSLATIONAL MEDICINE", ITMS: 26220220021 supported by the Operational Programme Research and Innovation funded by the ERDF.
Competing interests
All authors declare no conflict of interest.
References
- Edmondson, R., Broglie, J. J., Adcock, A. F. and Yang, L. (2014). Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol 12(4): 207-218.
- Johnson, S., Chen, H., and Lo, P. (2013). In vitro tumorsphere formation assays. Bio-protocol 3(3): e325.
- Pampaloni, F., Reynaud, E. G. and Stelzer, E. H. (2007). The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol 8(10): 839-845.
- R Core Team (2015). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. (https://www.R-project.org/)
- Strnadel, J., Choi, S., Fujimura, K., Wang, H., Zhang, W., Wyse, M., Wright, T., Gross, E., Peinado, C., Park, H. W., Bui, J., Kelber, J., Bouvet, M., Guan, K. L. and Klemke, R. L. (2017). eIF5A-PEAK1 signaling regulates YAP1/TAZ protein expression and pancreatic cancer cell growth. Cancer Res 77(8): 1997-2007.
- Weiswald, L. B., Bellet, D. and Dangles-Marie, V. (2015). Spherical cancer models in tumor biology. Neoplasia 17(1): 1-15.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Strnadel, J., Woo, S. M., Choi, S., Wang, H., Grendar, M. and Fujimura, K. (2018). 3D Culture Protocol for Testing Gene Knockdown Efficiency and Cell Line Derivation. Bio-protocol 8(11): e2874. DOI: 10.21769/BioProtoc.2874.
Category
Cancer Biology > Cancer stem cell > Cell biology assays > Cell isolation and culture
Cell Biology > Cell signaling > Intracellular Signaling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.