- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Modified Low-quantity RNA-Seq Method for Microbial Community and Diversity Analysis Using Small Subunit Ribosomal RNA

Published: Vol 8, Iss 9, May 5, 2018 DOI: 10.21769/BioProtoc.2828 Views: 6167
Reviewed by: Dennis NürnbergStefan de VriesShyam Solanki
Original research article
The authors used this protocol in:

Oct 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
We propose a modified RNA-Seq method for small subunit ribosomal RNA (SSU rRNA)-based microbial community analysis that depends on the direct ligation of a 5’ adaptor to RNA before reverse-transcription. The method requires only a low-input quantity of RNA (10-100 ng) and does not require a DNA removal step. Using this method, we could obtain more 16S rRNA sequences of the same regions (variable regions V1-V2) without the interference of DNA in order to analyze OTU (operational taxonomic unit)-based microbial communities and diversity. The generated SSU rRNA sequences are also suitable for the coverage evaluation for bacterial universal primer 8F (Escherichia coli position 8 to 27), which is commonly used for bacterial 16S rRNA gene amplification. The modified RNA-Seq method will be useful to determine potentially active microbial community structures and diversity for various environmental samples, and will also be useful for identifying novel microbial taxa.
Keywords: RNA-SeqBackground
Ribosomal RNA (rRNA) accounts for more than 90% of the total microbial RNA, and is suitable for the analysis of microbial communities as an indicator of microbial physiological activity to synthesize proteins (Blazewicz et al., 2013). The study of microbial community transcripts, including rRNA and mRNA, in a particular environment (Double RNA metatranscriptomics) has advantages in providing both functional and taxonomic information on microbes (Urich et al., 2008), but has failed to perform OTU-based community comparisons. Although diversity indices could be calculated and compared using the V3 region of 16S rRNA sequences when gel-extracted SSU rRNA is analyzed, only a third of the resulting 16S rRNA sequences were found to be suitable for such analyses (Li et al., 2014). Besides, such method usually requires high quantities of RNA (Li et al., 2014). We recently developed a modified RNA-Seq method that uses an immediate adaptor ligation step at the 5’ end of the RNA prior to reverse transcription. Consequently, we can obtain more 16S RNA reads which can be used for OTU-based community and diversity analysis especially regarding low-RNA-yield samples such as tap water, shower curtain and human skin (Yan et al., 2017), and also mudflat sediment samples (Yan et al., 2018).
Materials and Reagents
- RNA extraction
- RNA-Seq library preparation
- PCR tubes (Corning, Axygen®, catalog number: PCR-02-L-C )
- RNA-Seq Library Preparation Kit for Whole Transcriptome Discovery–Illumina Compatible (Gnomegen, catalog number: K02421-T )
- DNase/RNase free water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9932 )
- QubitTM RNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32855 )
- RNaseOUTTM Recombinant Ribonuclease Inhibitor (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10777019 )
- QubitTM dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32854 )
- Gnome Size Selector (Gnomegen, catalog number: R02424S )
- PCR tubes (Corning, Axygen®, catalog number: PCR-02-L-C )
- Other materials
- Pipette tips (10 μl, 200 μl, 1,000 μl) (Corning, Axygen®, catalog numbers: TF-300-R-S , TF-200-R-S , TF-1000-R-S )
- 1.5 ml Eppendorf tubes (Eppendorf, catalog number: 022363204 )
- 100% ethanol
- RNase-free 70% ethanol
- Agarose (Biowest, catalog number: 111860 )
- 50x TAE Buffer (Sangon Biotech, catalog number: B548101-0500 )
- DL2,000 DNA Marker (Takara Bio, catalog number: 3427A )
- Pipette tips (10 μl, 200 μl, 1,000 μl) (Corning, Axygen®, catalog numbers: TF-300-R-S , TF-200-R-S , TF-1000-R-S )
Equipment
- Pipettes (0-10 μl, 10-100 μl, 100-1,000 μl) (Eppendorf, catalog numbers: k03030 , k03031 , k03032 )
- Vortex-genieTM 2 (QIAGEN, model: Vortex-Genie® 2 )
- Fresco 17 centrifuge (Thermo Fisher Scientific, Thermo ScientificTM, model: HeraeusTM FrescoTM 17 )
- Qubit 2.0 Fluorometer (Thermo Fisher Scientific, model: Qubit 2.0 )
- Automated thermal cycler TP600 (Takara Bio, model: TP600 )
- MagneSphere® Technology Magnetic Separation Stand (twelve-position) (Promega, catalog number: Z5342 )
- Electrophoresis apparatus (Bio-Rad Laboratories)
- Gel imaging system
Software
- Sickle v1.33 (Joshi and Fass, 2011)
- Mothur v1.33.3 (Schloss et al., 2009)
- QIIME v1.8.0 (Caporaso et al., 2010)
- MIPE (Zou et al., 2017)
Procedure
Note: Details of sample collection, storage and transport for low-biomass samples (tap water, shower curtain, mudflat water, leaf surface and human skin) please see the method described by Yan et al. (2017). Regarding mudflat sediment samples, details of sample collection and nucleic acid extraction are described in our recent work (Yan et al., 2018).
- Nucleic acid extraction
- Extract total RNA or total nucleic acids from low-biomass samples, which are unsuitable for DNase I digestion, using a mirVANATM miRNA Isolation Kit. Five or more swabs are preferred to be pooled for nucleic acid extraction, while a whole piece of filter membrane is preferred to be cut into small pieces (~0.5 mm2) for extraction. To completely disrupt microbial cell walls, vortex such preprocessed samples with 0.7 g glass beads and 0.7 ml Lysis/Binding Buffer in a 2 ml bead beating tube at maximum speed prior to extraction. After centrifugation at 13,000 x g for 5 min at 4 °C, transfer the supernatant to a new 1.5 ml tube and extract total RNA strictly following the manufacturer’s instructions.
- Visualize the RNA samples in a 1% (w/v) agarose gel after electrophoresis to assess the RNA integrity (Figure 1). Store total RNA or total nucleic acids at -80 °C. Quantify total RNA using a QubitTM RNA HS Assay Kit on Qubit 2.0 fluorometer before the preparation of RNA-Seq libraries.
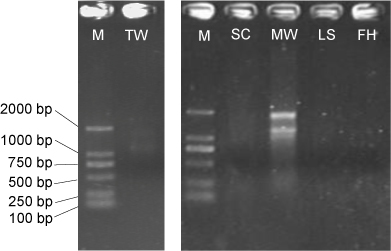
Figure 1. Total RNA electrophoresis for low-biomass samples. M: DL2,000 DNA marker; TW: tap water; SC; shower curtain; MW: mudflat surface water; LS: leaf surface; FH: forehead of human skin. Only TW and MW display integral 23S and 16S rRNA bands when 10 μl RNA is loaded, in contrast to samples that yield a very low concentration of RNA (< 2 ng/μl).
- Extract total RNA or total nucleic acids from low-biomass samples, which are unsuitable for DNase I digestion, using a mirVANATM miRNA Isolation Kit. Five or more swabs are preferred to be pooled for nucleic acid extraction, while a whole piece of filter membrane is preferred to be cut into small pieces (~0.5 mm2) for extraction. To completely disrupt microbial cell walls, vortex such preprocessed samples with 0.7 g glass beads and 0.7 ml Lysis/Binding Buffer in a 2 ml bead beating tube at maximum speed prior to extraction. After centrifugation at 13,000 x g for 5 min at 4 °C, transfer the supernatant to a new 1.5 ml tube and extract total RNA strictly following the manufacturer’s instructions.
- Preparation of RNA-Seq libraries
This RNA-Seq library preparation protocol is modified from the Gnomegen RNA-Seq Library Preparation Kit protocol (catalog number: K02421-T). Prepare RNA-Seq libraries following the steps described below.- Total RNA denaturation
- Add total RNA (10-100 ng) and DNase/RNase free water into a 0.2 ml PCR tube to make a 20-μl reaction volume.
- Mix well and incubate at 65 °C for 5 min using a thermal cycler program. Place the tube on ice immediately after the incubation and leave for at least 5 min.
Note: We use this step to substitute the original fragmentation step as to keep the integrity of the RNA and directly ligate the RNA-Seq adaptor to the 5’ end of the rRNA in the following step.
- Add total RNA (10-100 ng) and DNase/RNase free water into a 0.2 ml PCR tube to make a 20-μl reaction volume.
- Adaptor ligation to 5’ end of RNA
- Add the following reagents sequentially to the denatured RNA, and make a reaction volume of 40.6 μl:
10x ligation buffer 3.6 μl (Final ~0.9x) 10 mM ATP 4 μl (Final ~1 mM) Ligation enzyme mix 1 μl Ligation enzyme supplement 1 μl RNaseOUT (40 U/μl) 1 μl (Final ~1 U/μl) Ligation enhancer mix 8 μl RNA Seq 5’ Adaptor B 2 μl - Mix well and incubate at 37 °C for 2 h using a thermal cycler program. Place the tube on ice after incubation.
Note: RNaseOUT is a separately-purchased reagent. Such specific ligation between single-stranded Adaptor B and RNA avoids the interference of DNA for the community analysis.
- Add the following reagents sequentially to the denatured RNA, and make a reaction volume of 40.6 μl:
- Purification of ligation product
Transfer 40 μl ligation product to a new 1.5 ml tube, and purify the product using a Gnome Size Selector, strictly following the manufacturer’s instructions. A total volume of 10-μl purified product is obtained for the next step.
Note: Warm Gnome Size Selector to room temperature, and completely resuspend the Size Selector before use. Accurately use the volume that is recommended. - Synthesize the first strand of cDNA with a tagged random hexamer.
- Denaturation of ligation product
- Add 1 μl tagged RT primer and 1 μl dNTP (10 mM) to the purified ligation product in a 12-μl denaturation volume.
- Mix well and incubate at 65 °C for 5 min using a thermal cycler program. Place the tube on ice immediately after the incubation and leave for at least 5 min.
- Add 1 μl tagged RT primer and 1 μl dNTP (10 mM) to the purified ligation product in a 12-μl denaturation volume.
- Reverse transcription
- Add the following reagents sequentially into the denatured ligation product, and make a final 20-μl reaction volume:
5x First Strand Buffer 4 μl 100 mM DTT 2 μl RNaseOUT (40 U/μl) 1 μl Reverse Transcriptase 1 μl - Mix well and use the following thermal cycler program to synthesize the cDNA:
25 °C for 12 min;
42 °C for 40 min;
70 °C for 15 min;
Hold at 4 °C.
- Add the following reagents sequentially into the denatured ligation product, and make a final 20-μl reaction volume:
- Denaturation of ligation product
- cDNA purification
Purification is performed using a Gnome Size Selector strictly according to the manufacturer’s instructions. 20 μl purified cDNA is obtained for the next step.
Note: Warm Gnome Size Selector to room temperature, and completely resuspend the Size Selector before use. Accurately use the volume that is recommended. - PCR amplification
The complete reagent mixture contains:
Note: Forward and reverse primers are designed based on the RNA Seq 5’ Adaptor B and tag sequences, and are complementary to the standard Illumina forward and reverse primers. Therefore, the whole process avoids the utilization of microbe-related universal primers. The reverse primer also contains an 8-nucleotide (nt) indexing sequence to allow for multiplexing.Purified single-stranded cDNA 20 μl 2x HiFi PCR Master Mix 25 μl PCR Primer F 1.25 μl Barcoded reverse primer bcX 1.25 μl DNAse/RNase free water 2.5 μl
The PCR amplification conditions are as follows:
98 °C for 45 sec;
15 cycles of: 98 °C for 15 sec, 60 °C for 30 sec, 72 °C for 30 sec;
72 °C for 1 min;
Hold at 4 °C.
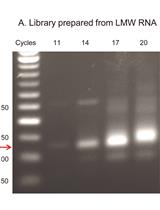
Note: Usually, 1-2 μl PCR products is enough for visualization on a 1% agarose gel. As a result, a wide range of clear and smear bands (100-2,000 bp) will be observed. - Purification of PCR product
Size select the 400-600 base pair (bp) PCR product using a Gnome Size Selector strictly according to the manufacturer’s instructions.
Note: Warm Gnome Size Selector to room temperature, and completely resuspend the Size Selector before use. Accurately use the volume that is recommended. Other size ranges of PCR product can also be obtained using Gnome Size Selector according to the manufacturer’s instructions.
- Total RNA denaturation
- High-throughput sequencing
Quantify barcoded PCR product from different samples using a QubitTM dsDNA HS Assay Kit on Qubit 2.0 fluorometer. Mix together PCR product from different samples with equal quantity (at least 10 ng for each sample) and sequence on an Illumina MiSeq platform using the 2 x 300 paired end protocol. For each sample, paired-end reads formatted as two FASTQ files will be obtained.
Data analysis
- Pre-process paired-end reads with Sickle software v1.33 using the command pe to trim and filter reads with a Phred quality score below 20.
- De novo merge paired-end reads using the command join_paired_end.py with default parameters in QIIME v1.8.0.
- Transform FASTQ files into fasta format using the command fastq.info in mothur v1.33.3.
- Remove merged sequences containing ambiguous nucleotides and homopolymer lengths longer than eight nucleotides using the command screen.seqs in mothur v1.33.3.
- Remove merged sequences shorter than 250 bp using the command screen.seqs in mothur v1.33.3.
- Classify the reads against the SILVA SSU v119 database in MIPE (Zou et al., 2017) with a bootstrap cut-off of 80% and remove SSU rRNA reads identified as chloroplast, mitochondria or human. Mismatches of the primer 8F (5’-AGAGTTTGAT (C/T) (A/C) TGGCTCAG-3’) (Mao et al., 2012) in all the bacterial 16S rRNA sequences are also identified with this software (Table 1).
Table 1. Non-coverage rates of the bacterial universal primer 8F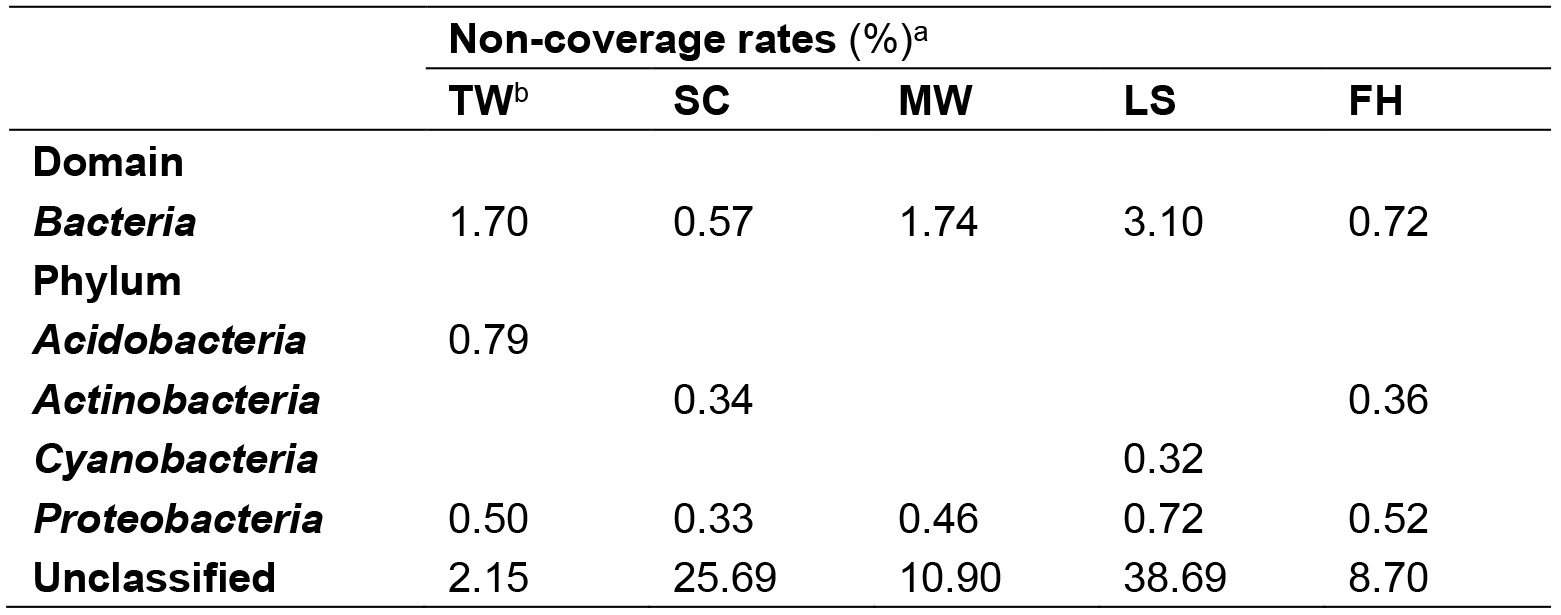
aThe non-coverage rates were calculated by dividing non-coverage sequences (at least one mismatch within primer 8F) of taxa with their relative total sequences. The phyla with less than 10 non-coveraged sequences in the datasets are not shown.
bTW: tap water; SC; shower curtain; MW: mudflat surface water; LS: leaf surface; FH: forehead of human skin. - For each sample, extract all the identified prokaryotic 16S rRNA reads using get.seqs command and deposit the reads in a fasta file. Merge all the fasta files of all the samples using cat command in Linux, and use count.seqs command in mothur v1.33.3 to differentiate sequences according to samples.
- Align and trim sequences to leave the 8F-V1-V2 regions (E. coli position 8 to 242) using align.seqs and pcr.seqs commands in mothur v1.33.3 respectively.
- Analyze OTU based community structures (Figure 2) and calculate diversity indices following the MiSeq SOP pipeline (https://www.mothur.org/wiki/MiSeq_SOP) as described in Kozich et al. (2013). Representative OTU sequences at a cut-off of 0.03 are taxonomically classified against the SILVA SSU v119 database with a bootstrap cut-off of 50%. OTUs belonging to chloroplasts or mitochondria are also removed from the analysis.
Note: All scripts for computational analysis are available in our supplementary material “Supplementary.docx”.
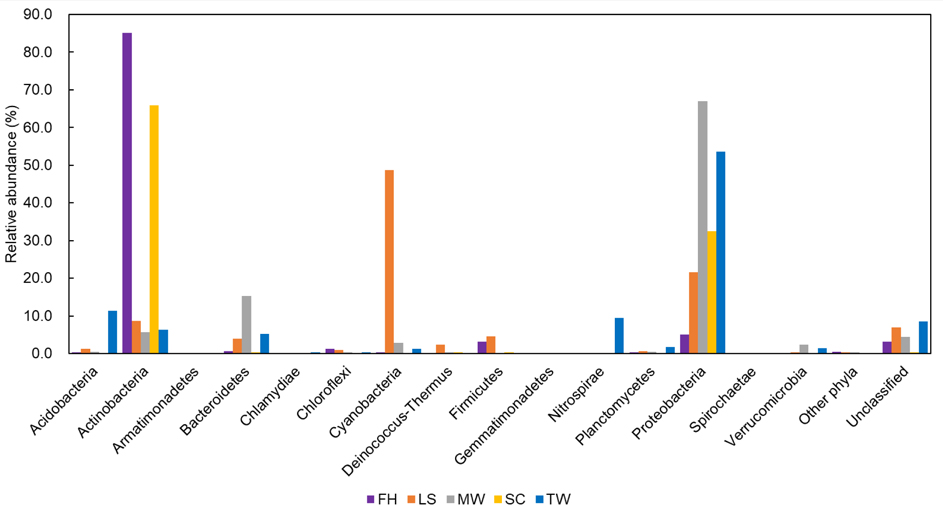
Figure 2. Comparisons of OTU-based bacterial communities in different samples at the phylum level. ‘Other phyla' includes taxa that made up of small fractions (< 1%). ‘Unclassified’ includes sequences under a bootstrap cut-off value of 50% for bacteria. TW: tap water; SC; shower curtain; MW: mudflat surface water; LS: leaf surface; FH: forehead of human skin.
Notes
- All the RNA samples and reaction reagents should be thawed and operated on ice. Once the reaction mix is prepared, perform the thermal cycler program as soon as possible.
- For different samples, the optimum cycles used in the PCR amplification step can be determined using a 10-μl preliminary reaction system.
Acknowledgments
This protocol was adopted from our previous study (Yan et al., 2017). This work was supported by the National Natural Science Foundation of China (NSFC) [grant number 31170114]. We declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Blazewicz, S. J., Barnard, R. L., Daly, R. A. and Firestone, M. K. (2013). Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J 7(11): 2061-2068.
- Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., Fierer, N., Pena, A. G., Goodrich, J. K., Gordon, J. I., Huttley, G. A., Kelley, S. T., Knights, D., Koenig, J. E., Ley, R. E., Lozupone, C. A., McDonald, D., Muegge, B. D., Pirrung, M., Reeder, J., Sevinsky, J. R., Turnbaugh, P. J., Walters, W. A., Widmann, J., Yatsunenko, T., Zaneveld, J. and Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5): 335-336.
- Joshi, N. A., and Fass, J. N. (2011). Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33).
- Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79(17): 5112-5120.
- Li, X. R., Lv, Y., Meng, H., Gu, J. D. and Quan, Z. X. (2014). Analysis of microbial diversity by pyrosequencing the small-subunit ribosomal RNA without PCR amplification. Appl Microbiol Biotechnol 98(8): 3777-3789.
- Mao, D. P., Zhou, Q., Chen, C. Y. and Quan, Z. X. (2012). Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC Microbiol 12: 66.
- Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. J. and Weber, C. F. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23): 7537-7541.
- Urich, T., Lanzen, A., Qi, J., Huson, D. H., Schleper, C. and Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One 3(6): e2527.
- Yan, Y. W., Jiang, Q. Y., Wang, J. G., Zhu, T., Zou, B., Qiu, Q. F. and Quan, Z. X. (2018). Microbial communities and diversities in mudflat sediments analyzed using a modified metatranscriptomic method. Front Microbiol 9: 93.
- Yan, Y. W., Zou, B., Zhu, T., Hozzein, W. N. and Quan, Z. X. (2017). Modified RNA-seq method for microbial community and diversity analysis using rRNA in different types of environmental samples. PLoS One 12(10): e0186161.
- Zou, B., Li, J., Zhou, Q. and Quan, Z. X. (2017). MIPE: A metagenome-based community structure explorer and SSU primer evaluation tool. PLoS One 12(3): e0174609.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yan, Y., Zhu, T., Zou, B. and Quan, Z. (2018). A Modified Low-quantity RNA-Seq Method for Microbial Community and Diversity Analysis Using Small Subunit Ribosomal RNA. Bio-protocol 8(9): e2828. DOI: 10.21769/BioProtoc.2828.
Category
Microbiology > Community analysis > RNA-Seq
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.