- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Electrophysiological Recordings of Evoked End-Plate Potential on Murine Neuro-muscular Synapse Preparations


(*contributed equally to this work) Published: Vol 8, Iss 8, Apr 20, 2018 DOI: 10.21769/BioProtoc.2803 Views: 13255
Reviewed by: Andrea PuharJackeline Moraes MalheirosAlexandros Kokotos
Original research article
The authors used this protocol in:

Aug 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Neuromuscular junction (NMJ) is the specialized chemical synapse that mediates the transmission of the electrical impulse running along motor neuron axons to skeletal muscle fibers. NMJ is the best characterized chemical synapse and its study along many years of research has provided most of the general knowledge of synapse development, structure and functionality.
Electrophysiology is the most accurate experimental procedure to study NMJ physiology and it largely contributed to the elucidation of synaptic transmission basic principles. Many electrophysiological techniques have been developed to study NMJ physiology and physiopathology. In this paper, we describe an ex vivo tissue preparation for electrophysiology that can be applied to investigate nerve-muscle transmission functionality in mice. It is routinely used in our laboratory to study presynaptic neurotoxins, antitoxins, and to monitor NMJ degeneration and regeneration. This is a broadly applicable technique which can also be adopted to investigate alterations of NMJ activity in mouse models of neuromuscular diseases, including peripheral neuropathies, motor neuron disorders and myasthenic syndromes.
Background
Neurotransmission is the physiological process by which neurons transfer information to target cells on a rapid time scale (usually < 1 msec). The structure mediating this communication is the synapse, a specialized structure formed either between neurons (a pre- and a post-synaptic neuron) or between a neuron (presynaptic neuron) and an effector cell (post-synaptic cell). Neuromuscular junction (NMJ) is the chemical synapse enabling communication between motor neuron and skeletal muscle fiber. This is the best characterized synapse and most of the knowledge on maturation, structure and function of synapses derives from its study (Li et al., 2016). At the NMJ, the action potential running along the motor axon invades the nerve terminal (presynaptic bouton) and induces the opening of voltage-gated calcium channels. The ensuing Ca2+ influx in presynaptic nerve terminal triggers (approximately in 0.3 µsec (Kuffler et al., 1984)) the fusion with the presynaptic membrane of about 100 synaptic vesicles from a ready to release pool (10-20% of all vesicles) (Del Castillo and Katz, 1954; Denker and Rizzoli, 2010). Around 1,000 acetylcholine (ACh) molecules per vesicle diffuse in the synaptic cleft (Kuffler and Yoshikami, 1975) and, in about 0.5 msec, bind to the nicotinic ACh Receptors (nAChRs) on the postsynaptic muscle fiber membrane. nAChRs are ionotropic ligand-gated Na+/K+ channels which open upon ACh binding and cause a local depolarizing potential of the postsynaptic membrane (end-plate) by mediating a large inward flux of Na+ (and a smaller outward flow of K+). This local depolarization is named evoked End-Plate Potential (eEPP) (or Evoked Junction Potential). In mice, the resting membrane potential of a skeletal muscle fiber lies around -75 mV and the eEPP has an amplitude of ~15-30 mV (depending on muscle type). When the eEPP amplitude is sufficiently high to reach or overcome action potential threshold, voltage-gated Na+ channels open thus triggering an action potential into the muscle fiber, which ultimately spreads out along the sarcolemma and invades muscle fiber T-tubules. Here, an excitation-contraction molecular machinery transduces this electric signal into the cytosolic release of Ca2+ from the sarcoplasmic reticulum, leading to muscle fiber contraction (Figure 1).
Figure 1. Mechanism of muscle fiber contraction. Acetylcholine (ACh), released by synaptic vesicle (SVs) fusing with the motor axon terminal membrane, binds to post synaptic nicotinic ACh Receptors, ionotropic cation channels that upon binding allow the leakage of cations (Na+ inward, K+ outward), leading to a local depolarization of the sarcolemmatic membrane (eEPP). When depolarization is sufficiently large to overcome the voltage threshold (red dotted line), voltage gated Na+ channels get open and trigger a post synaptic action potential (AP) spreading along the sarcolemma and invading the T-tubules (invagination of the sarcolemma within the muscle fiber). Dihydropyridine (DHP) receptors sense this membrane depolarization and stimulate the opening of Ryanodine Receptors (RyR) on the sarcoplasmic reticulum which release Ca++ into the cytosol eliciting muscle contraction. µ-conotoxin inhibits voltage gated Na+ channels thus allowing to record membrane depolarization due to the opening of the sole nicotinic AChR, i.e., the eEPP.
Random fusion of synaptic vesicles also takes place in the absence of a presynaptic action potential thereby inducing a very small (~0.4 mV) depolarization of the end-plate, which is not sufficient to trigger muscle contraction. This spontaneous activity is called ‘miniature End-Plate Potential’ (mEPP) and, according to the quantal hypothesis, it is generated by the release of a single synaptic vesicle (Katz, 2003).
NMJ is easily accessible to many kinds of experimental manipulation. Since the ‘50s’ electrophysiology applied at the NMJ provided seminal discoveries on basic aspects of synaptic transmission (Augustine and Kasai, 2007). Thereafter, a continuous development in technics and animal models paved the way to sophisticate investigation of pathological alterations occurring at the NMJ in neuromuscular diseases, including myasthenic syndromes and peripheral neuropathies, as well as neuroparalytic syndromes caused by animal (Duchen et al., 1981; Duregotti et al., 2015a) and bacterial toxins (Colasante et al., 2013, Pirazzini et al., 2014).
We describe here a detailed protocol to evaluate NMJ functionality in murine muscle-nerve preparations. The method is based on the intracellular recording of spontaneous mEPPs and nerve-evoked EPPs in muscle fibers of soleus nerve-muscle preparations, thus allowing accurate investigation of NMJ functionality at a single synapse resolution (Tremblay et al., 2017). We have recently used this method to test engineered botulinum neurotoxins and to assay the efficacy of novel putative antitoxins (Pirazzini et al., 2014; Zanetti et al., 2017). In addition, we successfully employed this technique to study NMJ nerve degeneration and to test molecules promoting its regeneration (Duregotti et al., 2015b; Negro et al., 2017; Rigoni and Montecucco, 2017).
This procedure represents a basic technique that can be easily adopted to investigate NMJ activity in mouse models of any neuromuscular diseases, including peripheral neuropathies, motor neuron disorders and myasthenic syndromes.
Materials and Reagents
- Silver wire (World Precision Instruments, catalog number: AGW2030 )
- 1 ml syringe (CHEMIL s.r.l., Padova, catalog number: S01G25 )
- Petri dish 35 mm (any producer is fine)
- Petri dish, 35 x 10 mm, coated with Sylgard (Dow Corning, Sylgard® 184 Silicone Elastomer kit)
- Flexible needle electrode Microfil (World Precision Instruments, catalog number: MF34G-5 )
- Tips for 2 μl micropipette
- Tips for 200 μl micropipette
- Tips for 1,000 μl micropipette
- Glass capillaries for intracellular microelectrodes (length 100 mm, inner diameter 0.86 mm, outer diameter 1.50 mm; Science Products, catalog number: GB150F-10 )
- Glass capillaries for stimulating microelectrode (length 100 mm, inner diameter 1.05 mm, outer diameter 1.50 mm; Science Products, catalog number: GB150TF-10 )
- Mice of desired strain and age
Note: We used here plp-GFP C57BL/6J transgenic mice. - Iron(III) chloride (FeCl3) (Sigma-Aldrich, catalog number: 451649 )
- Silver chloride (AgCl) (Sigma-Aldrich, catalog number: 204382 )
- μ-Conotoxin GIIIB (Alomone, Jerusalem, Israel)
- Sodium bicarbonate (NaHCO3) (Sigma-Aldrich, catalog number: S5761 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9333 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5655 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014 )
- Magnesium chloride, standard solution 1 M (MgCl2) (Honeywell International, Fluka, catalog number: 63020 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C5080 )
- Hydrogen chloride (HCl) (Sigma-Aldrich, catalog number: H1758 )
- Potassium acetate (CH3COOK) (Sigma-Aldrich, catalog number: P3542 )
- Ringer’s solution (see Recipes)
- Recording electrode solution (see Recipes)
Equipment
- Micropipettes
- Volumetric flask (typically 500 ml; any producer is fine)
- Electrophysiology setup complete with antivibration table (Newport, USA) (Figure 3)
- Stereomicroscope for the electrophysiology setup (Leica Microsystem, model: Leica MZ125 , numeric aperture 0.8 with plan apochromatic objective 1.6x) (#1 in Figure 3)
- Hydraulic micromanipulator for intracellular recording electrode (NARISHIGE, model: MHW-103 , Three-axis Water Hydraulic Micromanipulator) (#2 in Figure 3)
- Stimulation electrode micromanipulators (Manual Micromanipulator, MÄRZHÄUSER WETZLAR, model: MM 33 ) (#3 in Figure 3)
- Faraday cage (#4 in Figure 3, home-made) and stimulator: S88 stimulator (Grass, Warwick, RI, USA) (#5 in Figure 3)
- Amplifier: intracellular bridge mode amplifier (Npi electronic, model: BA-01X ) (#6 in Figure 3)
- 2 Forceps (Micro Jewelers Forceps, Rudolf Medical, catalog number: RU 4240-05 )
- Scissors (Micro Spring Scissors, Rudolf Medical, VANNAS, catalog number: RU 2260-08 )
- Scissors (Delicate Surgical Scissors, Rudolf Medical, catalog number: RU 1503-12 )
- Dissection microscope (OPTIKA Microscopes, model: SZM-LED2 )
- Pipette puller (P-97 Flaming/Brown Micropipette Puller) (Sutter Instruments, model: P-97 )
- A/D interface (National Instruments, model: PCI-6221 ) and computer compatible with the software (#7 in Figure 3)
- Gas tank 95% O2 with 5% CO2 (any size and any supplier are fine)
- Cylinder pressure regulator (Air Liquid, model: HBS 240-1-2 )
Software
- Recording: WinEDR free software (Strathclyde University, Glasgow, Scotland, UK)
- Analysis: Clampfit (Molecular Devices, Sunny-vale, CA, USA)
Procedure
- Solutions and setup preparation
- Pull the microelectrode for intracellular recording from filamented borosilicate glass (outer diameter: 1.5 mm; inner diameter 0.86 mm) and the microelectrode for nerve stimulation from filamented borosilicate glass (outer diameter: 1.5 mm; inner diameter 1.05 mm) using a pipette puller. Many electrodes can be pulled and stored at RT until use.
Note: The exact settings for pulling intracellular or stimulating microelectrodes may vary according to the glass (borosilicate vs. aluminosilicate), thickness of glass capillary wall, glass capillaries manufacturer and batch, puller heating lamina dimension, material and age, pulling velocity, time, steps, heat of each step, and also environmental conditions (humidity). These parameters are particularly relevant for intracellular microelectrodes and must be accurately set up until electrode resistance is optimal (see below). This requires a careful understanding of glass microelectrode behavior under heating conditions, and a clear understanding of puller function and response when settings are adjusted to new values. It is advisable to pull many capillaries with different puller settings until correct parameters are defined. Resistance can be measured by utilizing a specific intracellular amplifier circuit which displays the measured resistance according to Ohm’s law. A desirable resistance should be between 10 and 30 MΩ, values which substantially correspond to very small tips, in the order of 0.5 micrometer diameter (see Figure 2). Tip size of stimulating electrode is not crucial as it can be adjusted to nerve diameter by breaking it with gentle touches toward the bottom of the recording chamber (see Figure 2A). - Buffer the Ringer’s solution (Recipe 1) to pH 7.4 by bubbling with 95% O2, 5% CO2 for at least 15 min.
Note: About 10 ml of solution is needed for one standard experiment. - Prepare silver chloride wire. Formation of AgCl is achieved by chemical oxidation of the Ag-coated electrode in 10 mM FeCl3/HCl solution. Immerse the electrode in the solution for 10 sec.
Note: This step should be periodically repeated (about every 2-3 weeks) due to the scraping of AgCl layer upon repeated insertion into glass microelectrode. - Back-fill recording electrodes with recording electrode solution (Recipe 2). This step is carried out in two sequential steps:
- Prefill the electrode by heating (at mid-length) with a lighter for some seconds and by rapidly bathing the bottom into the solution. Leave the prefilled electrode horizontal to allow the solution flowing upwards to the tip by capillarity (Figure 2B). This takes about 30 min;
- Using 1 ml syringe and Electrode Microfil capillaries, add recording electrode solution until the electrode is almost filled (see Figure 2C).
Note: Avoid the injection of air bubbles during electrode filling.
Figure 2. Stimulation and recording electrodes. A. Stimulation electrode. Pre-filled (B) and filled (C) recording electrode. Arrows indicate the level of solution filling.
- Prefill the electrode by heating (at mid-length) with a lighter for some seconds and by rapidly bathing the bottom into the solution. Leave the prefilled electrode horizontal to allow the solution flowing upwards to the tip by capillarity (Figure 2B). This takes about 30 min;
- Switch on the components of the electrophysiological setup (Figure 3).

Figure 3. Electrophysiological setup components. 1) Stereomicroscope dedicated to the electrophysiology set up; 2) Intracellular electrode micromanipulator; 3) Stimulation electrode micromanipulator; 4) Faraday cage; 5) Stimulator; 6) Amplifier; and 7) Computer with WinEDR free software. - Connect recording (filled) and stimulation electrodes to their electrode-holders. Place a Petri dish filled with Ringer’s solution under the microscope and immerse the recording electrode tip into the solution. Switch on the amplifier and test the resistance. Optimal values are between 10 and 30 MΩ.
Notes:- Electrode resistance is indirectly proportional to tip diameter (the larger the tip, the lower the resistance). This relationship may be altered by experimental issues: i) incomplete filling or obstruction of the tip, due to the formation of salt microcrystals or the presence of micro-air bubbles, tend to increase the resistance. To restore suitable values, hold the electrode tip immersed in the Ringer’s solution for a few minutes: salts should dissolve and resistance decrease. If not, gently rub the tip of the electrode on the bottom of the chamber to cause ‘a controlled damage’ to the tip. Resistance should drop.
- A sudden increase of microelectrode resistance is generally due to tip obstruction by tissue debris (connective tissue or adipose tissue). In this case, the tip can be cleaned by passing large amounts of positive or negative currents (many amplifiers have an ‘electrode cleaner function’). Alternatively, rubbing the tip against the bottom of the chamber may also help. If the resistance does not drop and/or the recording trace is noisy, the electrode must be replaced. When resistance is below 10 MΩ or the tip is too wide (it has been damaged), the electrode must also be replaced.
- Electrode resistance is indirectly proportional to tip diameter (the larger the tip, the lower the resistance). This relationship may be altered by experimental issues: i) incomplete filling or obstruction of the tip, due to the formation of salt microcrystals or the presence of micro-air bubbles, tend to increase the resistance. To restore suitable values, hold the electrode tip immersed in the Ringer’s solution for a few minutes: salts should dissolve and resistance decrease. If not, gently rub the tip of the electrode on the bottom of the chamber to cause ‘a controlled damage’ to the tip. Resistance should drop.
- Switch on the stimulator and set the parameters for stimulation: for eEPPs, stimuli of 0.4 msec duration and frequency of 0.5 Hz are suitable.
Note: These parameters must be optimized to measure the NMJ functionality for two reasons: a) pulse step duration is minimal to prevent an excessive stimulus artifact; b) pulse step frequency is very low to prevent NMJ ‘fatigue’. - Run WinEDR free software.
- Pull the microelectrode for intracellular recording from filamented borosilicate glass (outer diameter: 1.5 mm; inner diameter 0.86 mm) and the microelectrode for nerve stimulation from filamented borosilicate glass (outer diameter: 1.5 mm; inner diameter 1.05 mm) using a pipette puller. Many electrodes can be pulled and stored at RT until use.
- Dissection of soleus muscle
- Euthanize the mouse according to your animal handling protocol.
Note: All procedures were performed in accordance with the Italian laws and policies (D.L. No. 26, 14th March 2014), with the guidelines established by the European Community Council Directive 2010/63/UE and approved by the veterinary services of the University of Padova (O.P.B.A.-Organismo Preposto al Benessere degli Animali) (protocol 359/2015). All the procedures should be utilized according to the ethical standards of the Institution where experiments are carried out. - Use the scissors to cut the skin all around the ankle. With the mouse belly up cut along the thigh (Figure 4A).
- With the aid of a tweezer, skin hind limb muscles. Put the mouse back in supine position and block the paw with a pin. Using a small scissors, cut the Achilles tendon (Figure 4B).
- Gently, pull the tendon up and, with Vannas scissors, accompany muscle detachment from the peroneal bone by cutting the connective tissue lateral to the soleus muscle (Figure 4C).
Notes:- Electrophysiological Recordings of mEPPs and eEPPs can be virtually done in any muscle that is collectible without damaging muscle fibers and with its nerve properly attached. Here we provide the detailed procedure for the soleus muscle that is, in our opinion, the easiest one to harvest.
- Cut the connective tissue from the inside to the outside to avoid (soleus) muscle damaging.
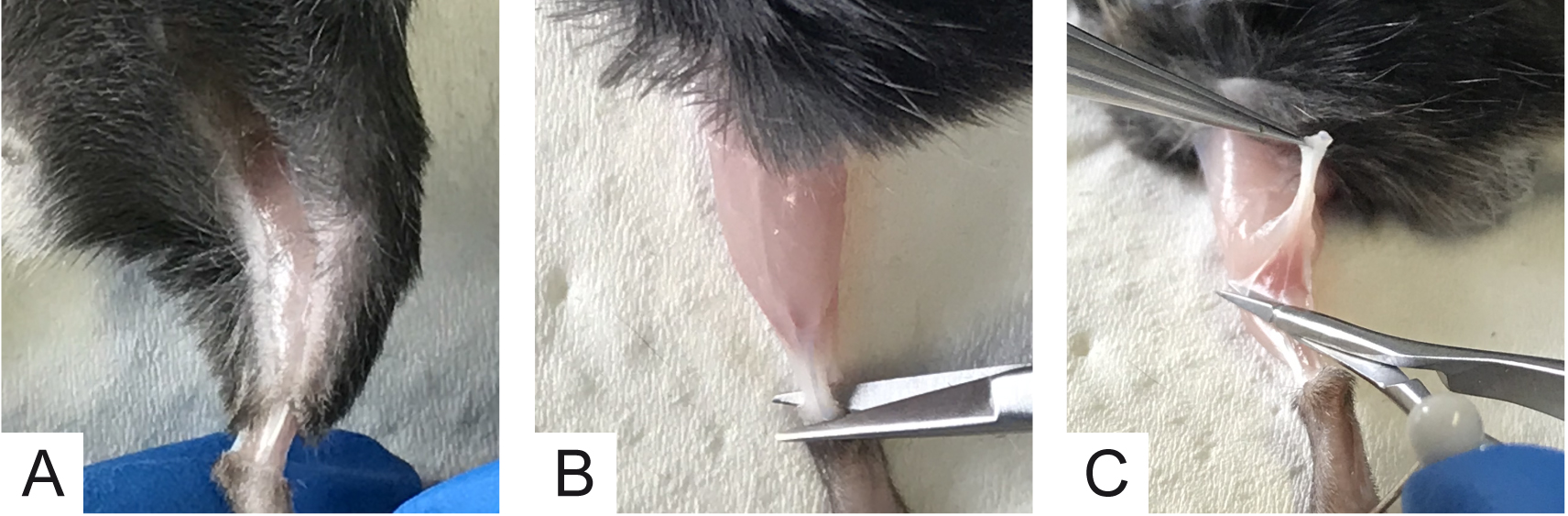
Figure 4. Initial steps for soleus muscle collection. A. Cut skin all around the ankle and along the thigh; B. Cut Achilles tendon; C. Arrows show the cut direction to detach connective tissue avoiding muscle damage. - Electrophysiological Recordings of mEPPs and eEPPs can be virtually done in any muscle that is collectible without damaging muscle fibers and with its nerve properly attached. Here we provide the detailed procedure for the soleus muscle that is, in our opinion, the easiest one to harvest.
- When the entire soleus muscle is visible, carefully sever the second tendon as close as possible to the knee, using small Vannas scissors (Figure 5A). At this point, the soleus muscle retires towards the Achilles tendon and all the muscles attached to the tendon can be excised without affecting soleus muscle integrity (Figure 5B).
- Place muscles in a Petri dish bottomed with sylgard and filled with oxygenated Ringer’s solution. Pin them as shown in Figure 5C, with the soleus muscle toward the operator (black arrow). Place the Petri dish under the stereomicroscope for next steps.
Note: For dissection, Ca2+ free Ringer’s solution is advisable to prevent muscle contraction or contracture due to mechanical stimulation.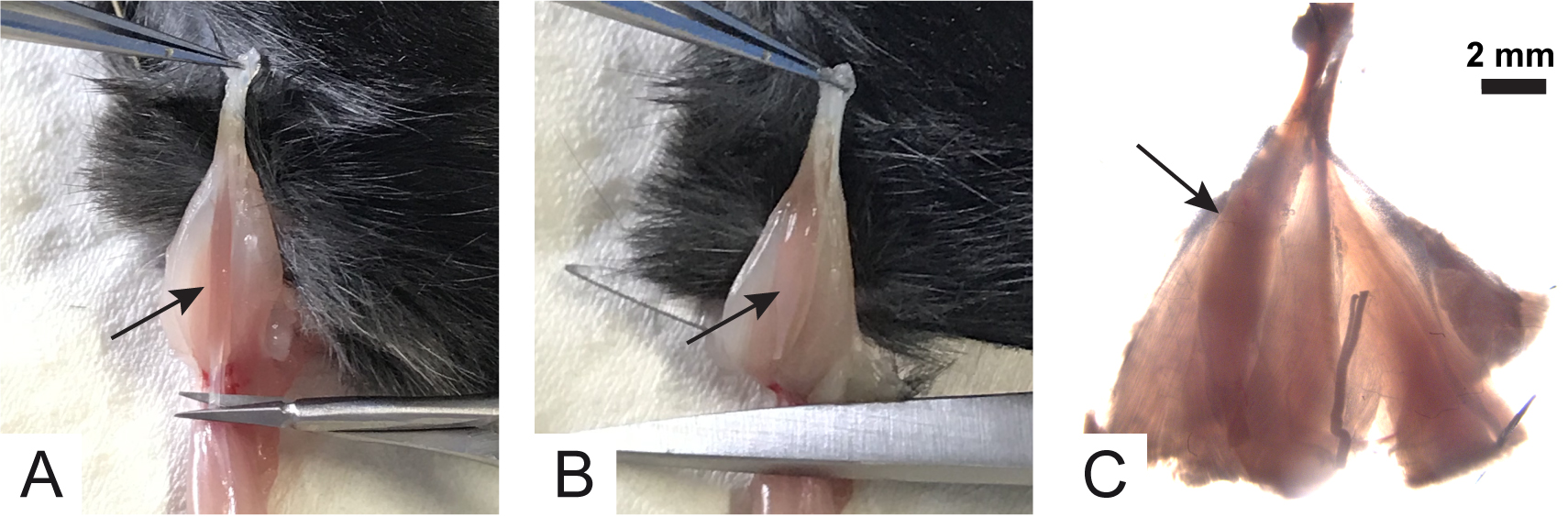
Figure 5. Collection of lower hind limb muscles. Arrows indicate soleus muscle. - Using forceps, gently lift the soleus muscle by grasping the lower tendon and by cutting the connective tissue that keeps it attached to the gastrocnemius muscle. Continue until the nerve is visible (Figure 6A). The nerve is underneath the soleus and looks like a white wire inserting into the muscle fibers (dotted line in Figures 6A-6C).
- Using Vannas scissors, clean up the nerve from the connective tissue (Figure 6B).
Note: Approximately 0.5 cm of nerve length is needed for optimal stimulation. - When the soleus muscle and its nerve are completely free, cut away the gastrocnemius muscle at the level of Achilles tendon (Figure 6C).
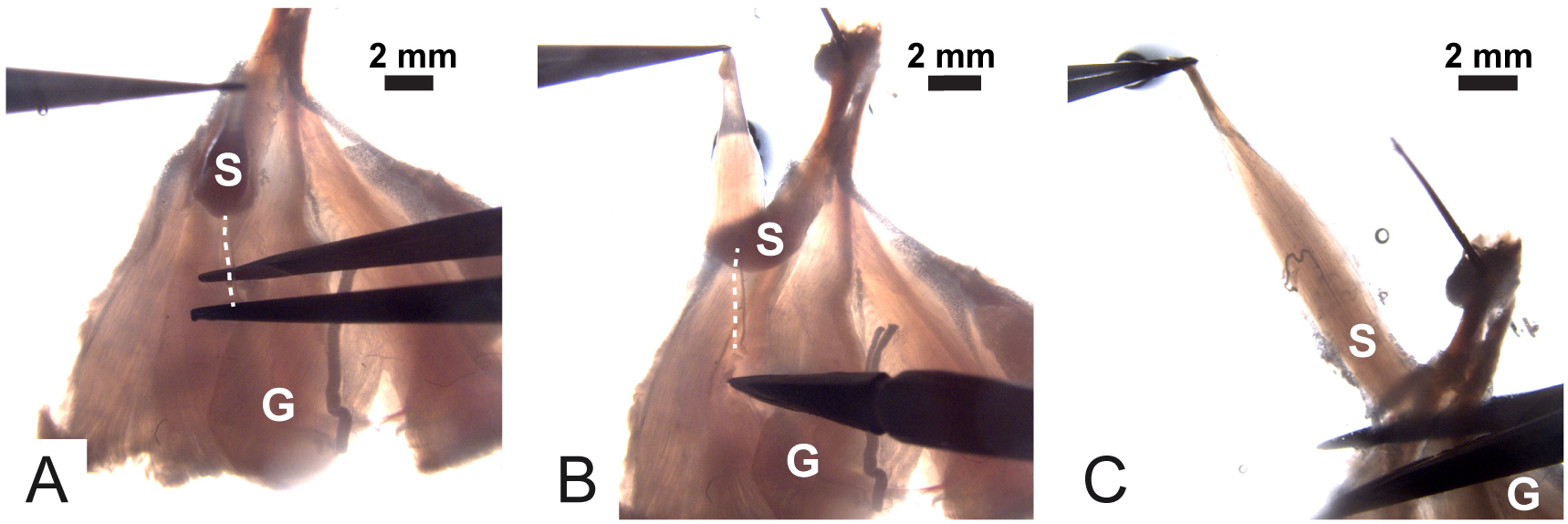
Figure 6. Isolation and dissection of the soleus with its nerve still associated. Dotted lines spot the nerve; S is the soleus muscle and G is the gastrocnemius muscle. - Pin the soleus to the bottom of the Petri dish via the two tendons (Figure 7A).
Note: The soleus muscles must be pinned in order to stretch the muscle and facilitate the entry of the recording electrode into the muscle fiber. Avoid excessive stretching which may cause damage to muscle fibers.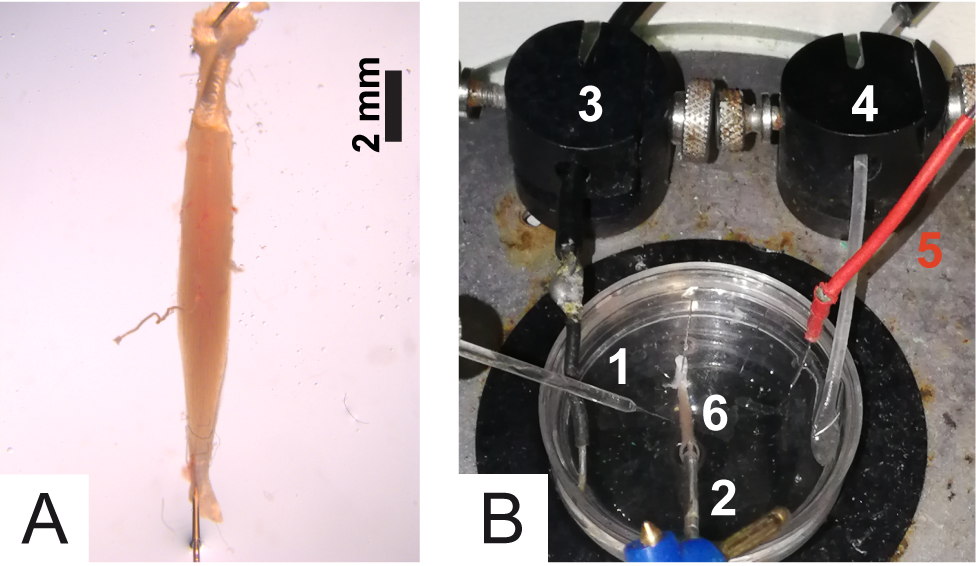
Figure 7. Details of the experimental chamber. A. Muscle positioning into the Petri dish; B. 1) Recording electrode; 2) Stimulation electrode; 3) Ground; 4) Tube for oxygenation; 5) Reference electrode; 6) Soleus muscle. - Gently, clear muscle and nerve from residual tissue debris without damaging the fibers or the nerve.
- Rinse muscle one or two times with Ringer’s solution to eliminate connective tissue debris or fur-hairs. Leave the muscle in the Petri dish filled with 2 ml of fresh Ringer’s solution.
- Euthanize the mouse according to your animal handling protocol.
- Nerve stimulations and EPPs recording
- Place the Petri dish under the stereomicroscope of the electrophysiology apparatus and submerge a tube connected to the O2/CO2 gas tank to bubble oxygen in the solution (Figure 7B).
- Put the Ground and the Reference of the stimulation electrode in the solution (Figure 7B). Note: Grounding is needed to get rid of electrical noise due to current loops among the apparatuses internal to the Faraday cage (Faraday cage, in turn, is necessary to prevent grid current noise). The Reference is needed to measure the voltage across the muscle fiber membrane, i.e., resting potential and how it varies (mEPPs, eEPPs and action potentials). Membrane potential is measured by comparing the voltage of the recording electrode with that of the Reference, which must be in contact with the extracellular fluid (solution) around the muscle.
- Using the micromanipulator, place the stimulation electrode in the solution and aspirate Ringer’s solution (filling approximately half of the electrode volume). Then, approach the electrode to the nerve stump and suck it up into the electrode together with the solution (Figure 8).
Note: Use a syringe to generate a negative pressure to aspirate the solution and to suck the nerve. The nerve must be completely immersed in the solution to avoid air entry into the electrode and to provide electrical continuity between the electrode and the nerve.
Figure 8. Positioning of electrodes. A. Recording electrode 1) Soleus muscle, 2) Recording electrode and 3) Nerve. B. Stimulation electrode. 1) Soleus muscle, 2) Recording electrode, 3) Nerve and 4) Recording electrode. - Stimulate the nerve by increasing the voltage intensity until the muscle contracts. For intracellular recordings set the stimulator at a voltage intensity which is 1.5-fold the value necessary to achieve muscle contraction.
Note: Muscle contraction is generally achieved with a voltage between 5 and 10 V. If contraction is not achieved within 15 V, the nerve can be pushed out and sucked again into the electrode. Alternatively, replace the pins to reduce muscle stretching. If contraction is still not visible, probably the nerve has been injured during the dissection. Avoid voltages above 20 V as muscle contraction may be triggered by direct stimulation of the muscle. - By using the intracellular electrode micromanipulator, place the tip of the recording electrode into the Ringer’s solution of the chamber and turn on the amplifier. Offset the voltage to 0. Move the electrode tip nearby the muscle fibers within the boxed area shown in Figure 9, where the nerve enters the muscle and NMJs are concentrated. When the tip of the electrode approaches the muscle fiber, a slight polarization of the voltage is observed (around -20 mV).
Note: Even though not strictly necessary, the use of a transgenic mouse with fluorescent reporters in motor neurons or Schwann cells coupled to an epifluorescence stereomicroscope helps in spotting NMJs more accurately.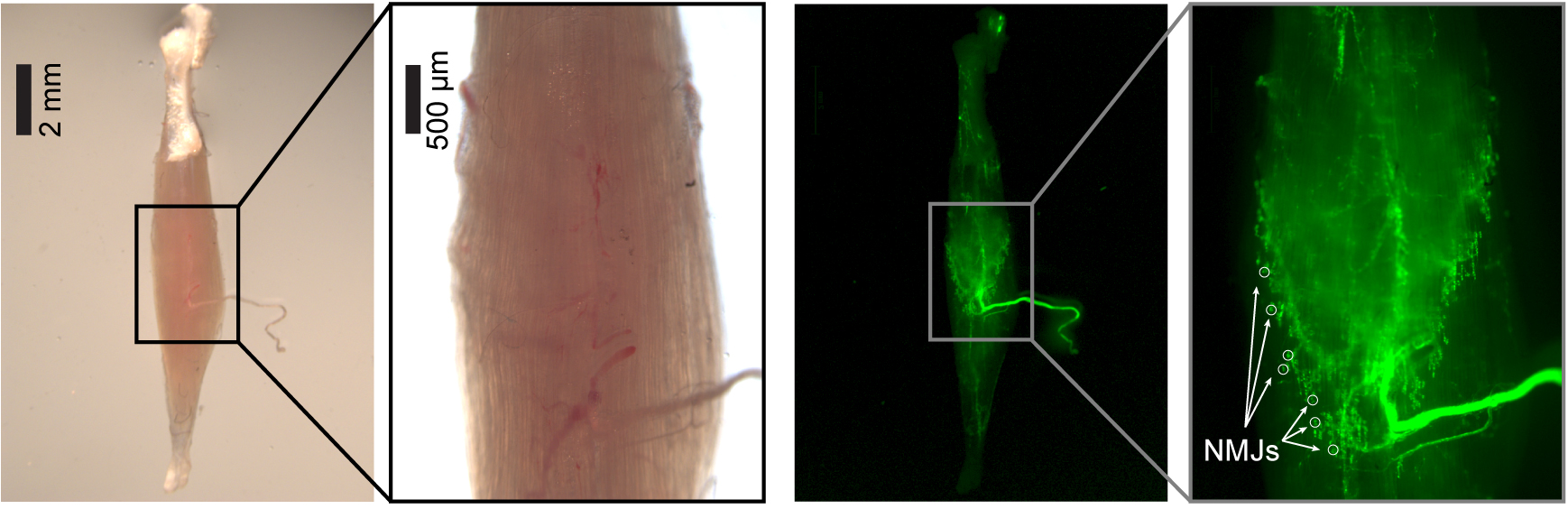
Figure 9. Distribution of NMJs. Soleus muscle from C57/BL6/J transgenic mice expressing GFP in Schwann cells under the plp promoter (plp-GFP (Mallon et al., 2002)), imaged with an epifluorescence stereomicroscope. With fluorescent Schwann cells, NMJs can be easily spotted at the end of nerve axons (white circles). - With the micromanipulator move the recording electrode deep toward the muscle. When a muscle fiber is correctly impaled, the voltage (read from the amplifier or the Oscilloscope/Computer interface) suddenly drops to very negative values, corresponding to the resting potential (VRest) of the muscle fiber (generally in between -90 and -60 mV).
Notes:- The value of the VRest varies from cell to cell and, in some case from the treatment. In this case, recordings are from a skeletal muscle cell where the VRest is between -60 and -90 mV. To obtain comparable eEPPs amplitudes (eEPP amplitude is linearly dependent on VRest (Boyd and Martin, 1955)) VRest is arbitrarily set to -70 mV for standardisation by using current injection commands.
- VRest is NEGATIVE. A positive potential likely corresponds to a misplacing of the electrode outside the fiber.
- If the resting potential is between -20 and -40 mV, the electrode is not properly inserted into the muscle cell. Amplifiers are generally equipped with the ‘BUZZ function’, a circuit based on oscillations caused by overcompensating the capacitance compensation system, which facilitates electrode entrance into the fiber.
- If membrane potential gradually depolarizes (i.e., slowly returns toward 0), muscle fiber membrane may be damaged and leaky or the electrode is getting out. Change fiber.
- Damaged fibers will not display the expected membrane potential in any case, thus change fiber.
- Prolonged denervation, as in the case of treatment with Botulinum neurotoxins or cut of nerves, increases the resting membrane potential. A common consequence is muscle fibrillation, a spontaneous, random and asynchronous contraction of muscle fibers (Purves and Sakmann, 1974; Moravec and Vyskocil, 2005). Fibrillation can be inhibited by specific drugs (see below).
- The value of the VRest varies from cell to cell and, in some case from the treatment. In this case, recordings are from a skeletal muscle cell where the VRest is between -60 and -90 mV. To obtain comparable eEPPs amplitudes (eEPP amplitude is linearly dependent on VRest (Boyd and Martin, 1955)) VRest is arbitrarily set to -70 mV for standardisation by using current injection commands.
- At this point, mEPPs can be recorded.
Note1: Upon stimulation, action potentials are elicited and can be recorded. Instead, eEPPs cannot be recorded yet as they are hidden/covered by action potentials.
Note2: Please consider that action potentials cause muscle contraction that may break the recording electrode or cause its exit from the fiber during the recording. - Add μ-Conotoxin GIIIB to selectively inhibit voltage-gated Na+ channels (Nav1.4 predominantly expressed by muscle) responsible for triggering the post synaptic action potential, thereby stopping muscle contraction. Using a micropipette, add 1 µl of μ-Conotoxin stock solution (1 mM) to the Petri dish, and switch on oxygenation.
- After 15 min, stimulate and check muscle contraction. If the muscle does not contract, EPPs can be recorded, otherwise wait additional 5-10 min.
Note: Deeper muscle fibers may need more time for μ-Conotoxin activity. - Stop oxygenation (solution movement due to bubbling or excessive flowing introduce artifacts to recording) and start the stimulation.
Note: eEPP amplitude is directly proportional to the amount of neurotransmitter release that in turn is directly proportional to the number of synaptic vesicles released upon stimulation. eEPPs can be therefore used to test the functional status of nerve endings, providing an accurate estimation of presynaptic effect. - Set recording time (at least 30 sec) and record eEPPs in current-clamp mode. Repeat the procedure for a dozen fibers per muscle (if eEPPs are heterogeneous within the same muscle, increase fiber sampling). A typical recording trace is shown in Figure 10, where the stimulation artifact (proportional to the amplitude of the stimulation), the EPP, proportional to nerve terminal activity and the mEPPs can be distinguished.
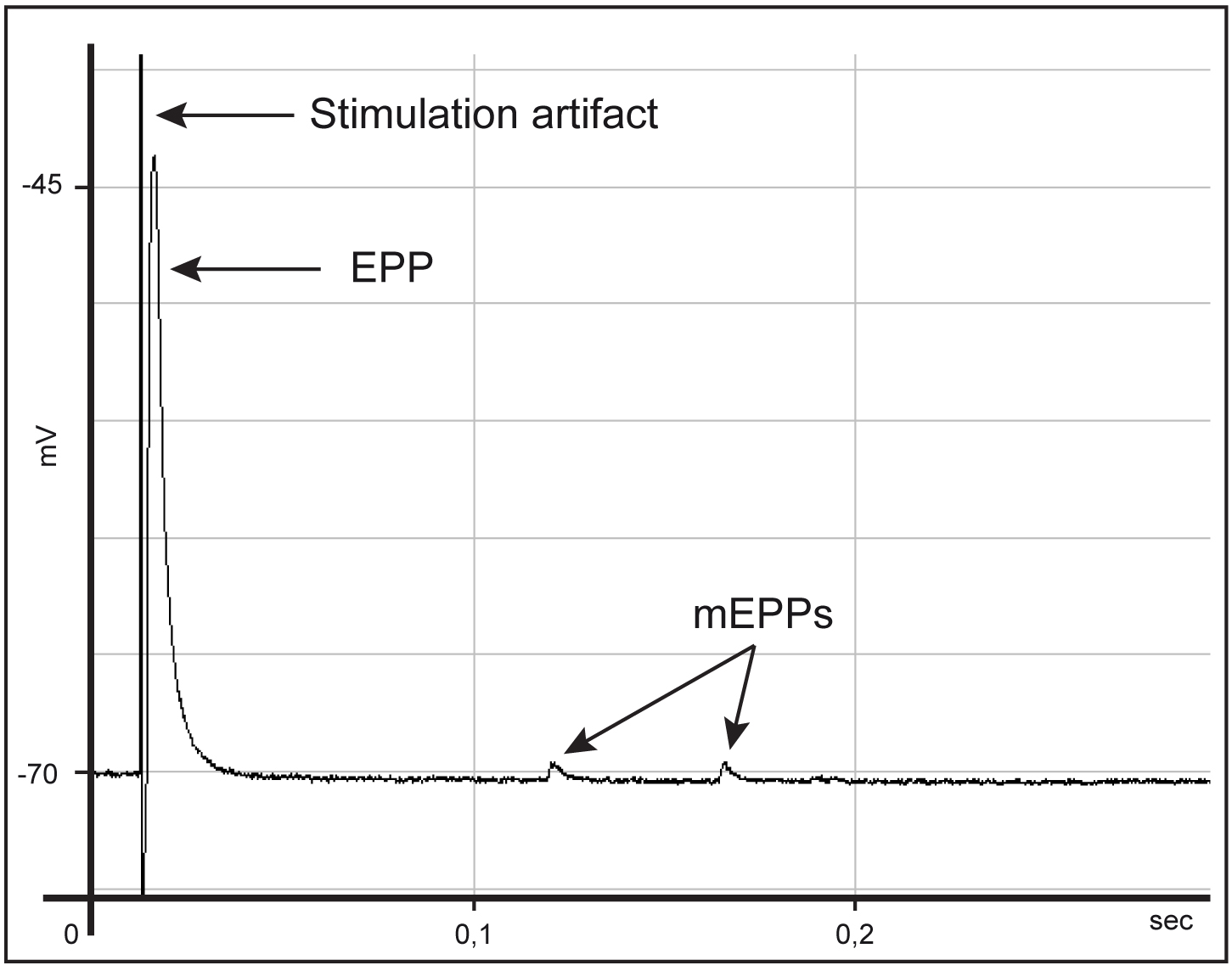
Figure 10. Typical trace during acquisition
- Place the Petri dish under the stereomicroscope of the electrophysiology apparatus and submerge a tube connected to the O2/CO2 gas tank to bubble oxygen in the solution (Figure 7B).
Data analysis
- The WinEDR software provides an EDR file format. Using WinEDR software, export this file as ABF file format compatible with Clampfit software.
Note: The procedure of data analysis may vary according to the software used for recording and analysis. - Open ABF file with Clampfit software and build a template on a control trace. Defining the shape of eEPPs in a control trace will ‘teach’ to Clampfit software to recognize the shape of eEPPs (or mEPPs) automatically in all other files. Analyse all the files of analyses to be compared with the same template. Clampfit software provides (for each event in the trace) a table with the parameters defining EPP form (start time, end time, peak amplitude, area etc.).
Note: The general shape of EPP is parabolic. The initial slope is related to the average amplitude of the postsynaptic response to a packet of transmitter (postsynaptic efficacy); the degree of curvature is related to the probability of transmitter release (presynaptic efficacy); the amplitude is related to the number of independent presynaptic release sites in the presynaptic terminal. In general, the ratio between eEPP and mEPP amplitudes is calculated as ‘quantal content’ of eEPP, i.e., the number of vesicles undergone fusion following a presynaptic action potential. Theoretically, a visual comparison of the different curves under different experimental conditions provides insights about what synaptic parameters are altered.
Recipes
- Ringer’s solution
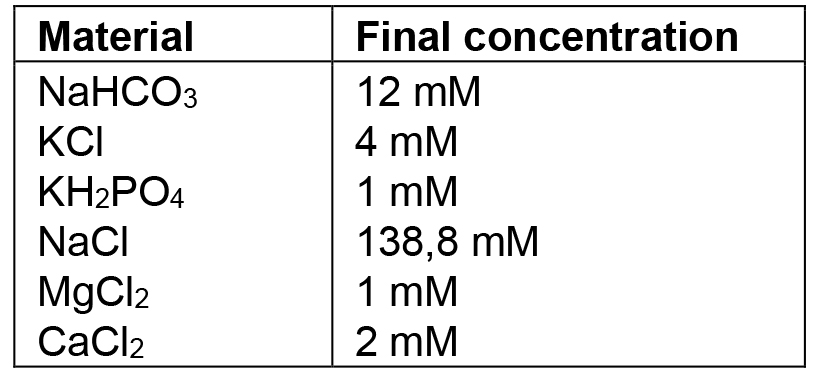
Notes:- Use ddH2O. All the glassware used must be washed with 0.1 M hydrogen chloride to remove any trace of carbonate, and rinsed very well. Once prepared, this solution can be stored at 4 °C, but no longer than 3 months. If deposit or opalescence is present, discard and prepare a fresh solution.
- Before the analysis, the Ringer’s solution is saturated with 95% O2, 5% CO2, by aeration for at least 15 min to obtain pH level of 7.4.
- Use ddH2O. All the glassware used must be washed with 0.1 M hydrogen chloride to remove any trace of carbonate, and rinsed very well. Once prepared, this solution can be stored at 4 °C, but no longer than 3 months. If deposit or opalescence is present, discard and prepare a fresh solution.
- Recording electrode solution

Acknowledgments
This work was supported by the University of Padova with ‘Senior Research Grant for young people not employed in the University of Padova’ granted to M. Pirazzini and with a ‘junior fellowship’ to S. Negro and by Fondazione Caritro with ‘Bando 2017 per giovani ricercatori coinvolti in progetti di eccellenza’ granted to G. Zanetti. All the procedures were performed in the laboratory of ‘Neurotoxins, Neuroparalysis and Regeneration’ headed by Prof. Cesare Montecucco at the Department of Biomedical Sciences (University of Padova). The authors declare no competing interests. SN and GZ performed the procedure for soleus muscle preparation with the help of AM. SN and GZ took the pictures to describe all the procedures and prepared the figures. GZ and MP wrote the paper, with the help of SN. All authors reviewed the manuscript and approved the final version.
References
- Augustine, G. J. and Kasai, H. (2007). Bernard Katz, quantal transmitter release and the foundations of presynaptic physiology. J Physiol 578(Pt 3): 623-625.
- Boyd, I. A. and Martin, A. R. (1955). The quantal composition of the mammalian end-plate potential. J Physiol 129(1): 14-15P.
- Colasante, C., Rossetto, O., Morbiato, L., Pirazzini, M., Molgo, J. and Montecucco, C. (2013). Botulinum neurotoxin type A is internalized and translocated from small synaptic vesicles at the neuromuscular junction. Mol Neurobiol 48(1): 120-127.
- Del Castillo, J. and Katz, B. (1954). Quantal components of the end plate potentials. J Physiol 124: 560-573.
- Denker, A. and Rizzoli, S. O. (2010). Synaptic vesicle pools: An update. Front Synaptic Neurosci 2: 135.
- Duchen, L. W., Gomez, S. and Queiroz, L. S. (1981). The neuromuscular junction of the mouse after black widow spider venom. J Physiol 316: 279-291.
- Duregotti, E., Negro, S., Scorzeto, M., Zornetta, I., Dickinson, B. C., Chang, C. J., Montecucco, C. and Rigoni, M. (2015a). Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc Natl Acad Sci U S A 112(5): E497-505.
- Duregotti, E., Zanetti, G., Scorzeto, M., Megighian, A., Montecucco, C., Pirazzini, M. and Rigoni, M. (2015b). Snake and spider toxins induce a rapid recovery of function of botulinum neurotoxin paralysed neuromuscular junction. Toxins (Basel) 7(12): 5322-5336.
- Katz, B. (2003). Neural transmitter release: from quantal secretion to exocytosis and beyond. J Neurocytol 32(5-8): 437-446.
- Kuffler, S. W., Nicholls, J. and Martin, R. A. (1984). From neuron to brain: A cellular approach to the function of the nervous system. Sinauer Associates, Sunderland, MA.
- Kuffler, S. W. and Yoshikami, D. (1975). The number of transmitter molecules in a quantum: an estimate from iontophoretic application of acetylcholine at the neuromuscular synapse. J Physiol 251(2): 465-482.
- Li, L., Xiong, W. C. and Mei, L. (2017). Neuromuscular junction formation, aging, and disorders. Annu Rev Physiol 80: 159-188.
- Mallon, B. S., Shick, H. E., Kidd, G. J. and Macklin, W. B. (2002). Proteolipid promoter activity distinguishes two populations of NG2-positive cells throughout neonatal cortical development. J Neurosci 22(3): 876-885.
- Moravec, J. and Vyskocil, F. (2005). Early postdenervation depolarization develops faster at endplates of hibernating golden hamsters where spontaneous quantal and non-quantal acetylcholine release is very small. Neurosci Res 51(1): 25-29.
- Negro, S., Lessi, F., Duregotti, E., Aretini, P., La Ferla, M., Franceschi, S., Menicagli, M., Bergamin, E., Radice, E., Thelen, M., Megighian, A., Pirazzini, M., Mazzanti, C. M., Rigoni, M. and Montecucco, C. (2017). CXCL12α/SDF-1 from perisynaptic Schwann cells promotes regeneration of injured motor axon terminals. EMBO Mol Med 9(8): 1000-1010.
- Pirazzini, M., Azarnia Tehran, D., Zanetti, G., Megighian, A., Scorzeto, M., Fillo, S., Shone, C. C., Binz, T., Rossetto, O., Lista, F. and Montecucco, C. (2014). Thioredoxin and its reductase are present on synaptic vesicles, and their inhibition prevents the paralysis induced by botulinum neurotoxins. Cell Rep 8(6): 1870-1878.
- Purves, D. and Sakmann, B. (1974). Membrane properties underlying spontaneous activity of denervated muscle fibres. J Physiol 239(1): 125-153.
- Rigoni, M. and Montecucco, C. (2017). Animal models for studying motor axon terminal paralysis and recovery. J Neurochem 142 Suppl 2: 122-129.
- Tremblay, E., Martineau, E. and Robitaille, R. (2017). Opposite synaptic alterations at the neuromuscular junction in an ALS mouse model: when motor units matter. J Neurosci 37(37): 8901-8918.
- Zanetti, G., Sikorra, S., Rummel, A., Krez, N., Duregotti, E., Negro, S., Henke, T., Rossetto, O., Binz, T. and Pirazzini, M. (2017). Botulinum neurotoxin C mutants reveal different effects of syntaxin or SNAP-25 proteolysis on neuromuscular transmission. PLoS Pathog 13(8): e1006567.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zanetti, G., Negro, S., Megighian, A. and Pirazzini, M. (2018). Electrophysiological Recordings of Evoked End-Plate Potential on Murine Neuro-muscular Synapse Preparations. Bio-protocol 8(8): e2803. DOI: 10.21769/BioProtoc.2803.
Category
Neuroscience > Peripheral nervous system > Skeletal muscle
Cell Biology > Cell-based analysis > Electrophysiological technique
Cell Biology > Cell signaling > Synaptic transmision
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.