- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Design of Hybrid RNA Polymerase III Promoters for Efficient CRISPR-Cas9 Function
Published: Vol 8, Iss 6, Mar 20, 2018 DOI: 10.21769/BioProtoc.2779 Views: 9921
Reviewed by: David CisnerosMichael TschernerSadri Znaidi
Original research article
The authors used this protocol in:

Apr 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The discovery of the CRISPR-Cas9 system from Streptococcus pyogenes has allowed the development of genome engineering tools in a variety of organisms. A frequent limitation in CRISPR-Cas9 function is adequate expression levels of sgRNA. This protocol provides a strategy to construct hybrid RNA polymerase III (Pol III) promoters that facilitate high expression of sgRNA and improved CRISPR-Cas9 function. We provide selection criteria of Pol III promoters, efficient promoter construction methods, and a sample screening technique to test the efficiency of the hybrid promoters. A hybrid promoter system developed for Yarrowia lipolytica will serve as a model.
Keywords: Synthetic biologyBackground
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) is a collection of DNA sequences found in bacteria that contain snippets of viral DNA from previous exposures (Marraffini and Sontheimer, 2010). The snippets are referred to as spacer DNA, and they are flanked by short, repetitive palindromic sequences. Bacteria use these stored spacer sequences as a template to express RNA to recognize and attack specific viruses if they are exposed again. When combined with CRISPR-associated (Cas) proteins, CRISPR-Cas systems can recognize and cut foreign DNA or RNA, destroying the virus and protecting the host from repeated infections (Barrangou, 2013).
A specific CRISPR system, the type II CRISPR-Cas9 from Streptococcus pyogenes, has been modified into a simpler system for use in genomic editing. With this system, researchers are able to design specific single-guide RNA (sgRNA) sequences that are complementary to a 20 bp sequence of a gene of interest that has an upstream protospacer adjacent motif (PAM; ‘NGG’) (Jinek et al., 2012). When the designed sgRNA complexes with the Cas9 protein, the assembled ribonucleoprotein binds to and introduces a double strand break (DSB) in the target DNA sequence. In genome editing applications, this DSB is then repaired by a cell’s native repair mechanisms. In the absence of an introduced repair template, the nonhomologous end-joining DNA repair pathway is normally used to repair the break in most eukaryotes (Moore and Haber, 1996). Repair via nonhomologous end-joining frequently results in an indel mutation that causes a frameshift mutation and disrupts the gene’s function. The simple programmability of the sgRNA sequences allows for unprecedented precision in genomic edits. In addition, the portability of the CRISPR-Cas9 system has allowed precise genome editing and other applications in organisms where it was previously tedious or impossible (Mali et al., 2013; Wang et al., 2013; Lobs et al., 2017b; Schwartz et al., 2017b and 2017c). The efficiency of the CRISPR-Cas9 system has been shown to correlate with sgRNA expression (Hsu et al., 2013; Ryan et al., 2014; Yuen et al., 2017). Because of this, a range of strategies for sgRNA expression have been developed. RNA polymerase II promoters, which primarily serve to drive expression of mRNA, have been used because they are widely studied and offer a high degree of control over expression (Deaner et al., 2017). More commonly for CRISPR systems, RNA polymerase III (Pol III) promoters have been used to drive sgRNA expression. Pol III promoters natively drive expression of smaller RNAs, most notably tRNAs, and yield higher transcript levels (Schwartz et al., 2016). To increase functional sgRNA expression levels even higher, Pol III promoters concatenated with tRNAs have been used (Xie et al., 2015; Schwartz et al., 2016; Lobs et al., 2017a). Implementation of synthetic hybrid Pol III promoter systems can improve CRISPR-Cas9 mediated genome editing for efficient gene disruption.
Materials and Reagents
- 10 µl pipette tips (Fisher Scientific, FisherbrandTM, catalog number: 02-707-438 )
- 200 µl pipette tips (Fisher Scientific, FisherbrandTM, catalog number: 02-707-417 )
- 1,000 µl pipette tips (Fisher Scientific, FisherbrandTM, catalog number: 02-707-403 )
- 1.5 ml microcentrifuge tubes (Fisher Scientific, FisherbrandTM, catalog number: 05-408-129 )
- 0.2 ml PCR tubes (Fisher Scientific, FisherbrandTM, catalog number: 14-230-215 )
- 100 x 15 mm Petri dishes (Fisher Scientific, FisherbrandTM, catalog number: FB0875712 )
- 14 ml culture tubes (Corning, Falcon®, catalog number: 352057 )
- Competent DH5α Escherichia coli (New England Biolabs, catalog number: C2987I )
- Yarrowia lipolytica strain Po1f (ATCC, catalog number: MYA-2613 )
- pCRISPRyl (Addgene, catalog number: 70007 ) or Episomal Cas9 plasmid (see Notes)
- Q5 HF polymerase (New England Biolabs, catalog number: M0491L )
- YeaStarTM Genomic DNA Kit (Zymo Research, catalog number: D2002 )
- DNA Clean & ConcentratorTM (Zymo Research, catalog number: D4004 )
- Gibson Assembly Master mix (New England Biolabs, catalog number: E2611L )
- ZyppyTM Plasmid Miniprep Kit (Zymo Research, catalog number: D4037 )
- CutSmart Buffer (New England Biolabs, catalog number: B7204S )
- AvrII restriction enzyme (New England Biolabs, catalog number: R0174S )
- Taq DNA polymerase (New England Biolabs, catalog number: M0273L )
- Yeast extract (BD, DifcoTM, catalog number: 212750 )
- Peptone (BD, DifcoTM, catalog number: 211677 )
- Glucose (Fisher Scientific, FisherbrandTM, catalog number: D16-10 )
- Agar (Sigma-Aldrich, catalog number: A7002-1KG )
- Yeast nitrogen base without amino acids (BD, DifcoTM, catalog number: 291940 )
- Complete Supplemental Mixture without Leucine (CSM-leu) (Sunrise Science, catalog number: 1005-010 )
- Complete Supplemental Mixture (CSM) (SunriseScience, catalog number: 1001-010 )
- Oleic acid (MP Biomedicals, catalog number: 0215178125 )
- Tween 20 (Sigma-Aldrich, catalog number: P9416-50ML )
- LB broth (Sigma-Aldrich, catalog number: L3022-1KG )
- Ampicillin (Sigma-Aldrich, catalog number: A0166 )
- YPD media/agar (see Recipes)
- SD-leu media/agar (see Recipes)
- SD oleic acid agar (see Recipes)
- LB agar (see Recipes)
Equipment
- Pipettes (Gilson, model: PIPETMANTM Variable Volume, catalog number: F167370 )
- Benchtop microcentrifuge (Eppendorf, model: 5424 , catalog number: 022620401)
- PCR thermocycler (Bio-Rad Laboratories, model: T100TM, catalog number: 1861096 )
- Incubation shaker (Infors, model: Multitron Standard )
- Incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: HerathermTM IGS60, catalog number: 51028063 )
- Gel electrophoresis tank (Bio-Rad Laboratories, model: Wide Mini-Sub®, catalog number: 1704468 )
- Gel electrophoresis power supply (Bio-Rad Laboratories, model: PowerPacTM Basic, catalog number: 1645050 )
- Gel imager (Bio-Rad Laboratories, model: Gel DocTM XR+, catalog number: 1708195 )
Procedure
- Promoter selection criteria
Our lab has previously developed CRISPR-Cas9 systems for the yeasts Y. lipolytica and Kluyveromyces marxianus (Schwartz et al., 2016; Lobs et al., 2017a). In these works, we compared CRISPR-Cas9 activity with sgRNA expression from native Pol III promoters, tRNAs, and hybrid Pol III promoters combining native Pol III promoters with a tRNA. In both organisms, hybrid Pol III promoters outperformed native Pol III promoters and tRNAs; however, the native Pol III promoter used in the best hybrid promoter was different in each organism. To date, only class II RNA Pol III promoters (those that are tRNA-like) have been demonstrated in hybrid promoters as described in this protocol. The class II promoters which can be tried include SNR52, SNR6, RPR1, and SCR1 (Marck et al., 2006). These can be identified in annotated genomes of an organism, or found via BLAST search using a closely related organism as input. In plant and mammalian systems, the Pol III U3 and U6 promoters have been extensively used for sgRNA expression, and so may be adaptable to a hybrid promoter approach (Cong et al., 2013; Mali et al., 2013; Shan et al., 2013). - Hybrid promoter construction
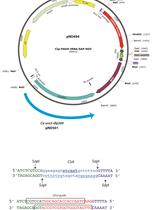
The hybrid promoter system combines the selected Pol III promoter and a tRNA sequence as shown in Figure 1. Sequences of tRNA from an organism can be identified via computational methods (Marck and Grosjean, 2002) or from a public database (http://gtrnadb.ucsc.edu/). Most often, such databases provide the mature tRNA sequence, which can be used to identify the full-length sequence in the genome via a BLAST search or a similar local alignment tool. In cases where the mature sequence is predicted or not know, experimental validation may be required. The addition of a tRNA allows the sgRNA to mature and be excised from the primary transcript. In addition, tRNAs are self-contained RNA Pol III promoters, which may result in improved sgRNA expression. The hybrid promoter consisting of the chosen RNA Pol III promoter and tRNA is placed upstream of the sgRNA encoding sequence with a polyT sequence for termination. This forms the complete hybrid promoter construct.
Figure 1. Synthetic hybrid promoter construction. A. Schematic of the assembly of Pol III promoter, tRNA, and AvrII-containing sgRNA via Gibson assembly. Similar colored boxes denote overlap sequences and the AvrII site. B. Schematic of cloning specific 20 bp sgRNA target sequence into digested AvrII site.- The desired Pol III promoter is amplified from genomic DNA in a PCR reaction with Q5 DNA polymerase. Genomic DNA can be extracted using an organism specific kit (for Y. lipolytica and K. marxianus we have used the YeaStarTM kit). Primers should be designed to bind ~200-300 bp upstream the ‘A-box’ and approximately 25 bp downstream from the ‘B-box’ of the Pol III promoter. The ‘B-box’ can be identified both by its putative consensus sequence (from Saccharomyces cerevisiae) ‘GWTCRAnnC’ and by its position downstream of an ‘A-box’ (consensus sequence ‘TRGYnnAnnnG’) (Marck et al., 2006). An example of a Pol III promoter amplified from genomic DNA is shown in Figure 2A. The primers used in the PCR reactions contain ~20-30 bp overlap sequences to join the truncated Pol III to the Cas9 plasmid backbone and the tRNA sequence via Gibson Assembly. The backbone homology sequence should contain a selected restriction site overhang. Example primers are shown below. The underlined sequences correspond to the promoter sequence of interest while the non-underlined sequences are the backbone and tRNA overlaps.
Forward primer:
CACATTTCCCCGAAAAGTGCCACCTGACGTCCCCAGTTGCAAAAGTTGACAC
Reverse primer:
AAACCATCGGCGCATTAGAGGTATTTTTCAAAGTAGCCCAGTGCAGAGTCC
Figure 2. Pol III promoter and tRNA design. A. Schematic of a Pol III promoter amplified from genomic DNA. The italicized dimensions are recommendations but will differ between organisms and between promoters. B. Schematic of a tRNA sequence amplified from genomic DNA. Similar to A, the italicized dimensions will differ between different tRNAs. The sequences that flank upstream the A-box and downstream the B-box are essential for Rnase P and Rnase Z binding and excision of the tRNA. - The tRNA sequence is similarly amplified from genomic DNA with Q5 DNA polymerase. Primers are designed ~20-30 bp upstream the A-box and ~20 bp downstream the B-box to allow for RNase P and RNase Z binding, respectively. Conserving these flanking sequences ensures excision of the matured tRNA. An example is seen in Figure 2B. The forward primer contains overlaps with the truncated promoter sequence. The reverse primer contains an overhang with an AvrII restriction site followed by an overlap of the sgRNA sequence. The AvrII restriction digestion site allows for easy addition of an N20 sgRNA targeting sequence. An example set of primers are shown below. The underlined sequence contains the tRNA sequence and the non-underlined sequences on the forward and reverse primer correspond to the truncated promoter and the AvrII (lower-cased)-sgRNA overlaps, respectively.
Forward primer:
CGAGTTCTGGACTCTGCACTGGGCTACTTTGAAAAATACCTCTAATGCGCCG
Reverse primer:
AACTTGCTATTTCTAGCTCTAAAAcctaggTCAACCTGCGCCGACCC - The final fragment containing the sgRNA sequence is amplified from plasmid pCRISPRyl (Addgene #70007) using primers containing AvrII- tRNA and Cas9 backbone overlaps. The underlined sequence contains the sgRNA sequence while the non-underlined sequences contain the AvrII(lower-cased)- tRNA overlap and plasmid backbone overlap with a restriction site overhang.
Forward primer:
CCGGTTCGATTCCGGGTCGGCGCAGGTTGAcctaggTTTTAGAGCTAGAAATAGCAAG
Reverse primer:
GTCATGATAATAATGGTTTCTTAGACGTAAAAAAAAGCACCGACTCGGTG - Once the Pol III, tRNA, and sgRNA sequences have been amplified and isolated using a PCR clean-up kit, like DNA Clean & ConcentratorTM, they are combined with the backbone plasmid in a single Gibson Assembly reaction as described below at a 1:3 ratio of backbone to insert. The backbone plasmid is cut with a restriction enzyme at a site distal to the Cas9 sequence. In pCRISPRyl, the AatII site is used.
- Gibson Assembly Conditions (10μl)
Incubate at 50 °C for 1 h.Gibson Master mix 5 μl Backbone plasmid 0.5 pmol Insert 1: Pol III fragment 1.5 pmol Insert 2: tRNA fragment 1.5 pmol Insert 3: sgRNA fragment 1.5 pmol H2O up to 10 μl
- Gibson Assembly Conditions (10μl)
- The product can then be directly transformed into DH5α competent cells for replication and extracted using ZyppyTM Plasmid Miniprep Kit.
- The desired Pol III promoter is amplified from genomic DNA in a PCR reaction with Q5 DNA polymerase. Genomic DNA can be extracted using an organism specific kit (for Y. lipolytica and K. marxianus we have used the YeaStarTM kit). Primers should be designed to bind ~200-300 bp upstream the ‘A-box’ and approximately 25 bp downstream from the ‘B-box’ of the Pol III promoter. The ‘B-box’ can be identified both by its putative consensus sequence (from Saccharomyces cerevisiae) ‘GWTCRAnnC’ and by its position downstream of an ‘A-box’ (consensus sequence ‘TRGYnnAnnnG’) (Marck et al., 2006). An example of a Pol III promoter amplified from genomic DNA is shown in Figure 2A. The primers used in the PCR reactions contain ~20-30 bp overlap sequences to join the truncated Pol III to the Cas9 plasmid backbone and the tRNA sequence via Gibson Assembly. The backbone homology sequence should contain a selected restriction site overhang. Example primers are shown below. The underlined sequences correspond to the promoter sequence of interest while the non-underlined sequences are the backbone and tRNA overlaps.
- sgRNA target sequence design
Selected sgRNA sequences can be designed into primers for cloning following the format below. The underlined segment is the desired N20 sgRNA sequence, the lowercase sequence is homologous to the AvrII restriction site while the rest of the sequence is overlap with the tRNA sequence and sgRNA. The overlaps allow sgRNA target sequence to be combined via Gibson Assembly into the AvrII digested CRISPR plasmid, as shown in Figure 1.
Forward primer:
GGGTCGGCGCAGGTTgacgtNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGC
Reverse primer:
GCTATTTCTAGCTCTAAAACNNNNNNNNNNNNNNNNNNNNacgtcAACCTGCGCCGACCC- The forward and reverse primers are annealed using the following protocol in a thermocycler to obtain a linear fragment.
- Annealing conditions (25 μl)
CutSmart buffer 2.5 μl Forward primer (10 μM) 5 μl Reverse primer (10 μM) 5 μl H2O 12.5 μl - Thermocycler protocol (7 min)
Continue decreasing in increments of 5 °C until the final temperature of 60 °C.95 °C 3 min 90 °C 30 sec 85 °C 30 sec
- Annealing conditions (25 μl)
- The backbone plasmid digested with AvrII as described below:
- Restriction digest conditions (50 μl)
CutSmart buffer 5 μl AvrII 1 μl Backbone plasmid 1 μg H2O up to 50 μl - Incubate at 37 °C for at least 1 h.
- Purify using DNA clean up kit.
- Restriction digest conditions (50 μl)
- Finally, the annealed fragment is combined with the backbone digested with AvrII via Gibson Assembly as described above at a ratio of 1:3 backbone to insert.
- The forward and reverse primers are annealed using the following protocol in a thermocycler to obtain a linear fragment.
- Measuring gene disruption efficiency
- Here, we describe an efficient screening method for verifying gene disruption in transformed colonies using PCR. This method can be used to simultaneously screen dozens of colonies.
- Isolate genomic DNA from random colonies after transformation using the YeaStar Genomic DNA Kit.
- Design primers that are 200 base pairs upstream and downstream of the sgRNA target sequence that will be used for amplification.
- Amplify the region flanking target site via PCR with Taq polymerase and purify.
- Sanger sequence the purified fragment and align with corresponding wild-type sequence to identify any indel or frameshift mutations.
- Isolate genomic DNA from random colonies after transformation using the YeaStar Genomic DNA Kit.
- Alternatively, quantification of gene disruption can be done with an easily selectable growth-associated phenotype. Here we use Y. lipolytica as an example. In Y. lipolytica, disruption of the gene PEX10 prevents peroxisome biogenesis and results in an inability to use long-chain fatty acids as an energy source (Blazeck et al., 2014; Schwartz et al., 2017a). This means that colonies with this phenotype are unable to grow on minimal media with oleic acid as the sole carbon source. Therefore, a CRISPR plasmid containing a sgRNA target sequence that targets PEX10 can be used to screen for genetic disruption, allowing simultaneous screening of hundreds of colonies.
- The wild-type strain of Y. lipolytica, PO1f, is transformed with the designed CRISPR plasmid at stationary phase (Schwartz et al., 2016). Successful transformants are selected for via outgrowth in either liquid SD-leu media or on SD-leu agar plates. Selection in SD-leu media ensures that all tested colonies contain the CRISPR plasmid. Outgrowth allows for expression of the CRISPR system and gene disruption before screening.
- Transformed colonies are randomly selected and streaked on both YPD and SD oleic acid agar plates and incubated at 30 °C.
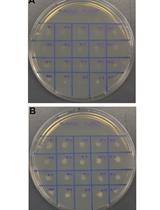
- Colonies that grow on YPD media but not SD oleic acid plates demonstrate genetic disruption of PEX10 as seen in Figure 3.

Figure 3. Phenotype of PEX10 disruptants. Example plates screening of PEX10 disrupted phenotypes on YPD and SD oleic acid media. PO1f is shown as a control, light blue indicates RPR1’-tRNAgly, orange indicates SNR52’-tRNAgly, and SCR1’-tRNAgly is shown in dark blue. Adapted with permission from Schwartz et al., 2016. Copyright © American Chemical Society 2015.
- The wild-type strain of Y. lipolytica, PO1f, is transformed with the designed CRISPR plasmid at stationary phase (Schwartz et al., 2016). Successful transformants are selected for via outgrowth in either liquid SD-leu media or on SD-leu agar plates. Selection in SD-leu media ensures that all tested colonies contain the CRISPR plasmid. Outgrowth allows for expression of the CRISPR system and gene disruption before screening.
- Here, we describe an efficient screening method for verifying gene disruption in transformed colonies using PCR. This method can be used to simultaneously screen dozens of colonies.
Data analysis
- Determining disruptions from sequence alignment
Alignment of amplified genomic sequences to the wild type may be done using a multiple sequence alignment program such as MUSCLE (https://www.ebi.ac.uk/Tools/msa/muscle/). This particular program allows for simultaneous alignment of up to 500 sequences. Aligning at least 50 bp sequences that span the target site allows for identification of indels. Indels of 1, 2, 4, and 5 bp (or any number not divisible by 3) indicate a frameshift mutation and a successful gene disruption. - Disruption efficiency
Disruption efficiency is measured in triplicate with each sample consisting of 30 randomly selected colonies. The percentage of disrupted colonies in each sample is calculated, and the mean disruption efficiency and standard deviation are determined and reported. For example, after 2 days of outgrowth, the synthetic hybrid promoter SCR1’-tRNAgly disrupted 15/30, 14/30, and 20/30 of colonies which gives a disruption efficiency of 54 ± 11% (Schwartz et al., 2016).
Notes
- Selection and design of an episomal Cas9 backbone plasmid is highly dependent on the organism. For example, pCRISPRyl used in our lab contains a Cas9 sequence that has been codon optimized for Y. lipolytica, a Y. lipolytica CEN sequence, and a Leucine selective marker. The plasmid also contains an ampicillin resistance cassette and origin of replication for propagation in E. coli. An analogous vector is needed for the organism of interest.
- The efficacy of selected tRNAs can differ between organisms, and so multiple different tRNA sequences may need to be tested in each case. Selection criteria include high native abundance and short length. In Y. lipolytica, tRNAgly was selected based on its high native abundance according to codon usage in the Y. lipolytica genome. Our particular tRNA sequence was the shortest tRNAgly.
- The PEX10 gene in Y. lipolytica allowed for an easily screened phenotype. However, while not all organisms can grow with oleic acid as a sole carbon source, other organisms may have similar growth-associated genes that allow for straight-forward screening methods. For example, the ADE2 gene in S. cerevisiae causes cells to appear red in the absence of adenine (Jones and Fink, 1982), while disruption of the XYL2 gene in K. marxianus eliminates its ability to grow on xylitol (Lobs et al., 2017a).
Recipes
- YPD media/agar
10 g/L yeast extract
20 g/L peptone
20 g/L glucose
For agar plates, add 20 g/L agar
Note: Glucose must be added after autoclaving and cooling to 50 °C. - SD-leu media/agar
7 g/L yeast nitrogen base without amino acids
0.69 g/L CSM-leu
20 g/L glucose
For agar plates, add 20 g/L agar
Note: Glucose must be added after autoclaving and cooling to 50 °C. - SD oleic acid agar
7 g/L yeast nitrogen base without amino acids
0.69 g/L CSM
20 g/L agar
0.3% oleic acid
0.2% Tween 20
Note: Oleic acid and Tween 20 must be added after autoclaving and cooling to 50 °C. - LB agar
20 g/L LB broth
15 g/L agar
Note: For selective plates add 50 μg/ml ampicillin after autoclaving and cooling to 50 °C.
Acknowledgments
This protocol was adapted from a previously published work (Schwartz et al., 2016) and supported by NSF CBET-1403264 and -1403099 and the University of California, Riverside Chancellor’s Research Fellowship. The authors declare no conflict of interest or competing interest.
References
- Barrangou, R. (2013). CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip Rev RNA 4(3): 267-278.
- Blazeck, J., Hill, A., Liu, L., Knight, R., Miller, J., Pan, A., Otoupal, P. and Alper, H. S. (2014). Harnessing Yarrowia lipolytica lipogenesis to create a platform for lipid and biofuel production. Nat Commun 5: 3131.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Deaner, M., Mejia, J., and Alper, H. (2017). Enabling graded and large-scale multiplex of desired genes using a dual-mode dCas9 activator in Saccharomyces cerevisiae. ACS Synth Biol 6(10) 1931-1943.
- Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., Li, Y., Fine, E. J., Wu, X., Shalem, O., Cradick, T. J., Marraffini, L. A., Bao, G. and Zhang, F. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9): 827-832.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Jones, E. W. and Fink, G. R. (1982). Regulation of amino acid and nucleotide biosynthesis in yeast. In: Strathern, J. N., Jones, E. W. and Broach, J. R. (Eds). The Molecular Biology of the Yeast Saccharomyces: Metabolism and Gene Expression. Cold Spring Harbor Laboratory Press pp: 191-299.
- Lobs, A. K., Engel, R., Schwartz, C., Flores, A. and Wheeldon, I. (2017a). CRISPR-Cas9-enabled genetic disruptions for understanding ethanol and ethyl acetate biosynthesis in Kluyveromyces marxianus. Biotechnol Biofuels 10: 164.
- Lobs, A. K., Schwartz, C. and Wheeldon, I. (2017b). Genome and metabolic engineering in non-conventional yeasts: Current advances and applications. Synth Syst Biotechnol 1-10.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Marck, C. and Grosjean, H. (2002). tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA 8(10): 1189-1232.
- Marck, C., Kachouri-Lafond, R., Lafontaine, I., Westhof, E., Dujon, B. and Grosjean, H. (2006). The RNA polymerase III-dependent family of genes in hemiascomycetes: comparative RNomics, decoding strategies, transcription and evolutionary implications. Nucleic Acids Res 34(6): 1816-1835.
- Marraffini, L. A. and Sontheimer, E. J. (2010). CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11(3): 181-190.
- Moore, J. K. and Haber, J. E. (1996). Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol 16(5): 2164-2173.
- Ryan, O. W., Skerker, J. M., Maurer, M. J., Li, X., Tsai, J. C., Poddar, S., Lee, M. E., DeLoache, W., Dueber, J. E., Arkin, A. P. and Cate, J. H. (2014). Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 3.
- Schwartz, C., Frogue, K., Misa, J. and Wheeldon, I. (2017a). Host and pathway engineering for enhanced lycopene biosynthesis in Yarrowia lipolytica. Front Microbiol 8.
- Schwartz, C., Frogue, K., Ramesh, A., Misa, J. and Wheeldon, I. (2017b). CRISPRi repression of nonhomologous end-joining for enhanced genome engineering via homologous recombination in Yarrowia lipolytica. Biotechnol Bioeng 114(12): 2896-2906.
- Schwartz, C. M., Hussain, M. S., Blenner, M. and Wheeldon, I. (2016). Synthetic RNA polymerase III promoters facilitate high-efficiency CRISPR-Cas9-mediated genome editing in Yarrowia lipolytica. ACS Synth Biol 5(4): 356-359.
- Schwartz, C., Shabbir-Hussain, M., Frogue, K., Blenner, M. and Wheeldon, I. (2017c). Standardized markerless gene integration for pathway engineering in Yarrowia lipolytica. ACS Synth Biol 6(3): 402-409.
- Shan, Q., Wang, Y., Li, J., Zhang, Y., Chen, K., Liang, Z., Zhang, K., Liu, J., Xi, J. J., Qiu, J. L. and Gao, C. (2013). Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31(8): 686-688.
- Wang, H., Yang, H., Shivalila, C. S., Dawlaty, M. M., Cheng, A. W., Zhang, F. and Jaenisch, R. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153(4): 910-918.
- Xie, K., Minkenberg, B. and Yang, Y. (2015). Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci U S A 112(11): 3570-3575.
- Yuen, G., Khan, F. J., Gao, S., Stommel, J. M., Batchelor, E., Wu, X. and Luo, J. (2017). CRISPR/Cas9-mediated gene knockout is insensitive to target copy number but is dependent on guide RNA potency and Cas9/sgRNA threshold expression level. Nucleic Acids Res 45(20): 12039-12053.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Misa, J., Schwartz, C. and Wheeldon, I. (2018). Design of Hybrid RNA Polymerase III Promoters for Efficient CRISPR-Cas9 Function. Bio-protocol 8(6): e2779. DOI: 10.21769/BioProtoc.2779.
Category
Molecular Biology > DNA > DNA modification
Microbiology > Microbial genetics > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.