- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Measuring Nucleosome Assembly Activity in vitro with the Nucleosome Assembly and Quantification (NAQ) Assay


Published: Vol 8, Iss 3, Feb 5, 2018 DOI: 10.21769/BioProtoc.2714 Views: 8809
Reviewed by: Gal HaimovichEmilia Krypotou Vinay Panwar
Original research article
The authors used this protocol in:

Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Nucleosomes organize the eukaryotic genome into chromatin. In cells, nucleosome assembly relies on the activity of histone chaperones, proteins with high binding affinity to histones. At least a subset of histone chaperones promotes histone deposition in vivo. However, it has been challenging to characterize this activity, due to the lack of quantitative assays.
Here we developed a quantitative nucleosome assembly (NAQ) assay to measure the amount of nucleosome formation in vitro. This assay relies on a Micrococcal nuclease (MNase) digestion step that yields DNA fragments protected by the deposited histone proteins. A subsequent run on the Bioanalyzer machine allows the accurate quantification of the fragments (length and amount), relative to a loading control. This allows us to measure nucleosome formation by following the signature DNA length of ~150 bp. This assay finally enables the characterization of the nucleosome assembly activity of different histone chaperones, a step forward in the understanding of the functional roles of these proteins in vivo.
Background
The eukaryotic genome is organized into nucleosomes. Nucleosomes are modular and dynamic structures composed of an octameric core of histone proteins, wrapped by 147 bp of DNA (Luger et al., 1997). Nucleosome assembly begins with the deposition of one (H3-H4)2 tetramer onto DNA to form a tetrasome. Subsequent incorporation of H2A-H2B dimers forms a hexasome, and finally the nucleosome. Histones are highly positively charged small proteins that primarily exist as histone dimers at physiological salt concentrations. Because of their charges, histones require chaperones which shuttle them from the cytoplasm to the nucleus, and then aid their deposition onto, or removal from DNA (Gurard-Levin et al., 2014).
Histone chaperones are grouped in families of structurally unrelated proteins, all characterized by high binding affinity for histones (Laskey et al., 1978). In this way, they shield the histone charges and prevent their non-specific interaction with DNA and other cellular factors (Elsässer and D’Arcy, 2013). How histone chaperones participate in these different roles, and the degree of division of labor among histone chaperones remain largely unknown.
This is due to the lack of mechanistic knowledge of histone chaperone function, in particular as histone deposition factors onto DNA. It is therefore critical to develop assays that can measure histone deposition activity, i.e., nucleosome assembly, to be able to fully understand the functions of this class of proteins and the dynamics of histones in cells.
Because histone chaperones are not enzymes per se, it has been challenging to develop reliable assays to measure their histone deposition activity. Most existing assays have used native gel analysis to assay nucleosome assembly (Muthurajan et al., 2016). The readout in these assays is prone to misinterpretation, as histones and DNA can form a variety of complexes and native gel analysis is not sufficient to accurately distinguish between the different histone-DNA complexes.
We have developed a nucleosome assembly and quantitation (NAQ) assay that measures the amount of nucleosome particles formation in vitro. This assay relies on the activity of Micrococcal Nuclease (MNase), an enzyme that digests DNA that is not bound by histone proteins. The subsequent purification of the DNA fragments provides a footprint of the histone-DNA complexes in solution. The characteristic protection of ~150 bp DNA is a signature of intact nucleosome species and can be measured using a Bioanalyzer apparatus (Muthurajan et al., 2016). Data normalization to a loading control DNA allows us to compare and accurately quantify the amount of nucleosomes formed in different samples. The NAQ assay has been successfully used to measure the activity of the chromatin assembly factor 1 (CAF-1) in vitro (Mattiroli et al., 2017a and 2017b), and has the potential to reveal the differential contribution of histone chaperones to nucleosome assembly in cells. This will pave the way for the complete understanding of their functional roles in nucleosome dynamics.
Materials and Reagents
- Low retention pipette tips (USA Scientific)
- PCR tubes (USA Scientific, catalog number: 1402-4308 )
- 1.5 ml tubes (Fisher Scientific, catalog number: 05-408-129 )
- 207 bp DNA (procedure explained in Dyer et al., 2004)
- Loading control DNA [of length between 400 and 1,000 bp, we use a 621 bp DNA (procedure explained in Muthurajan et al., 2016)]
- Refolded H2A-H2B (procedure explained in Dyer et al., 2004) or H2A-H2B labeled with ATTO 647N (procedure explained in Muthurajan et al., 2016).
- Refolded (H3-H4)2 (procedure explained in Dyer et al., 2004)
- Recombinant histone chaperone
- Salt-assembled nucleosome (procedures explained in Dyer et al., 2004)
- 50% glycerol (autoclaved and stored at room temperature)
- 50 bp DNA ladder (Gold Bio, catalog number: D100-500 )
- 10x MNase buffer (New England Biolabs, provided with M0247S )
- 100x BSA (New England Biolabs, catalog number: B9001 )
Note: This product has been discontinued. - Micrococcal nuclease (MNase) (New England Biolabs, catalog number: M0247S )
- MinElute kit (QIAGEN, catalog number: 28006 )
- Proteinase K, 20 mg/ml solution (BioExpress, catalog number: E195-5ML )
- Tris (2-Carboxyethyl) phosphine Hydrochloride (TCEP) (Gold Bio, catalog number: TCEP100 )
- Sodium hydroxide (NaOH) (Fisher Scientific, catalog number: S318-3 )
- Tris base (Fisher Scientific, catalog number: BP152-5 )
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: S271-10 )
- 500 mM EDTA solution at pH ~8 (stored at room temperature)
- Tween-20 (Fisher Scientific, catalog number: BP337 )
- SYBR Gold nucleic acid gel stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S11494 )
- Boric acid (Acros Organics, catalog number: 180570025 )
- Ammonium persulfate (AMRESCO, catalog number: 0486 )
- 30% acrylamide 37.5:1 (Life science Products, catalog number: EC-890 )
- Tetramethylethylenediamine (TEMED) (Fisher Scientific, catalog number: BP150-20 )
- Bromophenol blue (Fisher Scientific, catalog number: B392-5 )
- Xylene cyanol FF (Sigma Aldrich, catalog number: X4126 )
- Sodium acetate (Fisher Scientific, catalog number: S210-500 )
- Acetic acid (Avantor Performance Materials, catalog number: V193-46 )
- 1 M TCEP (Tris (2-Carboxyethyl) phosphine Hydrochloride; see Recipes)
- NA buffer (see Recipes)
- SYBR Gold stain solution (see Recipes)
- 10x TBE (Tris/Borate/EDTA; see Recipes)
- 25% APS (Ammonium Persulfate; see Recipes)
- 6% PAGE gels (see Recipes)
- 10% PAGE gels (see Recipes)
- DNA sample buffer (see Recipes)
- 3 M Na acetate pH 5.0 solution (see Recipes)
Equipment
- Pipettes (Gilson)
- PAGE running apparatus (Hoefer, model: SE250 )
- Thermoblock (Fisher Scientific, catalog number: 11-718 )
- Centrifuge (Eppendorf, model: 5417 C )
- Typhoon FLA 9500 (GE Healthcare, model: Typhoon FLA 9500, catalog number: 28996943 )
- Bioanalyzer (Agilent)
- DNA 1000 chip for Bioanalyzer (Agilent Technologies, catalog number: 5067-1504 )
Software
- Agilent Expert 2100 Software
- Excel Software
Procedure
The workflow of the NAQ assay is shown in Figure 1: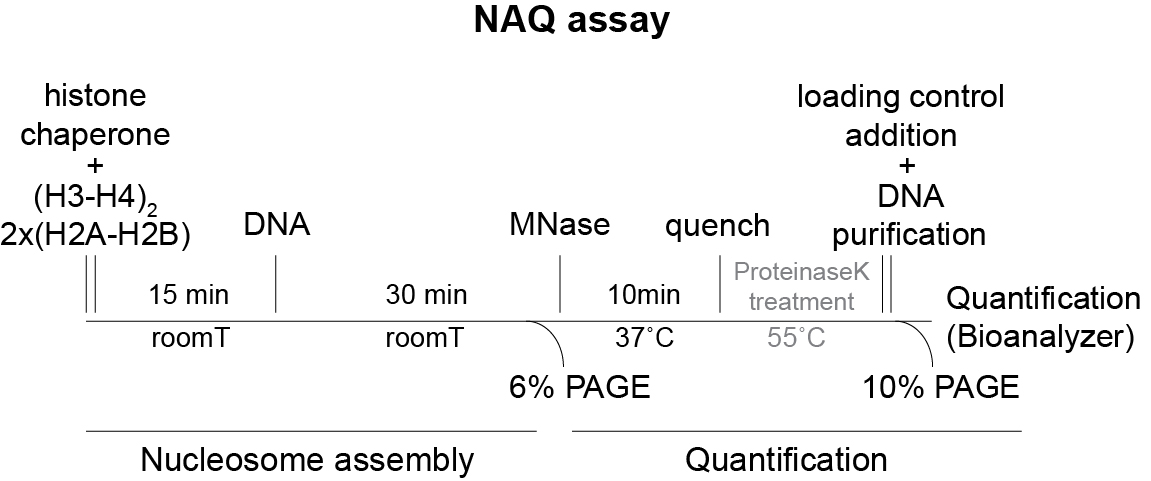
Figure 1. Workflow of the NAQ assay procedure. In gray are the optional steps.
- Nucleosome assembly reaction
- Prepare a 40 µl reaction in NA buffer (see Recipes) containing:
200 nM histone chaperone
200 nM (H3-H4)2 (tetramer concentration)
400 nM H2A-H2B (dimer concentration)
Notes:- It is important to pipette the histone chaperone first, as the histones are destabilized at low salt concentration.
- Histones are added subsequently in any order. Octamer preparations (Dyer et al., 2004) (200 nM) can also be used instead of separate (H3-H4)2 and H2A-H2B addition.
- Keep the stock solutions at 4 °C, but the reaction at room temperature (20 °C).
- We normally set up reactions for multiple histone chaperone concentrations, usually between 100 and 800 nM (or 0.5-4 times the concentration of histones and DNA components).
- Always include a ‘no chaperone’ reaction (where only histones are present), a chaperone only reaction (where no histones are present) and a salt-reconstituted nucleosome reaction (pre-assembled on 207 bp DNA [Dyer et al., 2004]). These are set up in parallel and treated exactly like the assembly reactions.
- It is important to pipette the histone chaperone first, as the histones are destabilized at low salt concentration.
- Incubate for 15 min at room temperature.
- Add 200 nM of 207 bp DNA, and mix by pipetting.
- Incubate for 30 min at room temperature.
- Proceed with the following steps and do not store the assembly reactions.
- Prepare a 40 µl reaction in NA buffer (see Recipes) containing:
- Native PAGE analysis of assembled nucleosomes
10 µl of the assembly reaction can be used for native PAGE analysis for validation purposes:- Add 2 µl of 50% glycerol stock.
- Load 4 µl of the final mix onto a 6% native PAGE (see Recipes) and run in 0.2x TBE buffer (pre-run gel at 150 V at 4 °C for 1 h, see Recipes).
- Include a lane with a DNA ladder (50 bp DNA ladder).
- Run samples at 150 V for 70 min at 4 °C.
- Stain with SYBR GOLD stain solution (see Recipes) for 20 min at room temperature.
- Image with Typhoon: Cy2/488 and A647 (if using ATTO 647N labeled H2A-H2B), with PMT (photomultiplier tube) at 500 V and resolution at 100 µm.
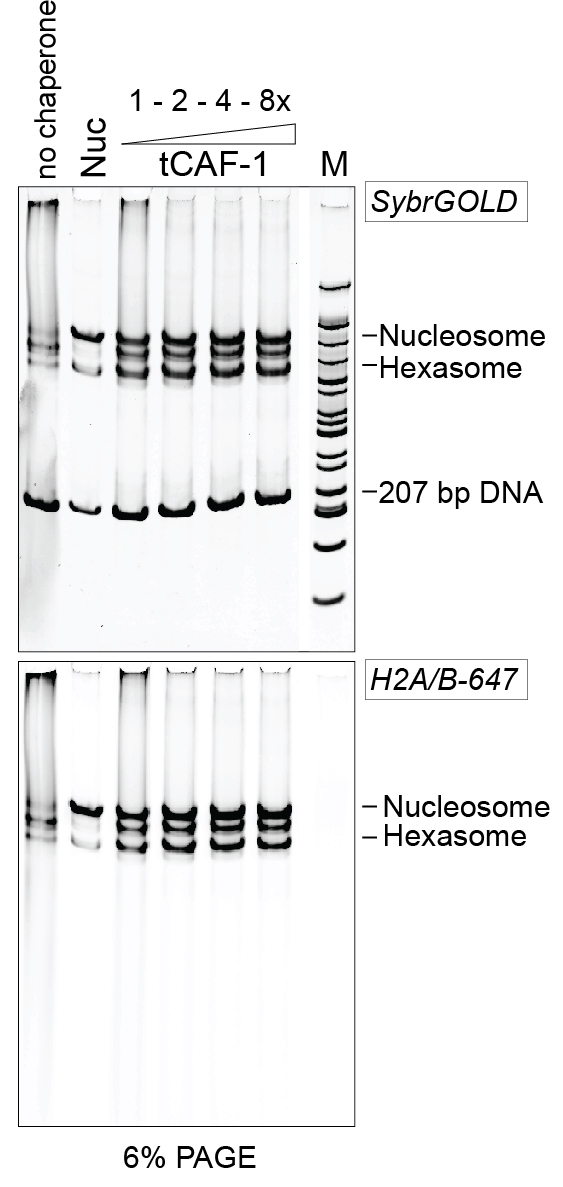
Figure 2. Example of 6% PAGE analysis of the purified DNA. Nuc stands for salt-assembled nucleosomes. tCAF-1 is an active construct of the histone chaperone CAF-1 (Mattiroli et al., 2017b). - Add 2 µl of 50% glycerol stock.
- Micrococcal Nuclease digestion and DNA purification
- Take 25 µl of the nucleosome assembly reaction and mix with 10 µl of 10x MNase buffer, 1 µl 100x BSA, 1 µl of MNase (stock at 25 U/µl) and 63 µl of water.
- Incubate for 10 min in a thermoblock at 37 °C
- Quench the reaction by adding 10 µl of 500 mM EDTA (final EDTA concentration ~50 mM).
- Optional: Treat the sample with 25 µg of Proteinase K (1.25 µl of a 20 mg/ml solution) for 20 min at 50 °C.
- Add 550 µl of PB buffer from MinElute kit (QIAGEN) and 10 µl of 3 M Na acetate pH 5.0 solution.
- Incubate for 10 min at room temperature.
- Add 50 ng of loading control DNA (stock at 25 ng/µl).
- Apply sample to the spin column.
- Centrifuge for 1 min at 16,000 x g at room temperature.
- Discard flow-through.
- Wash membrane with 100 µl of PB buffer.
- Centrifuge for 1 min at 16,000 x g at room temperature.
- Discard flow-through.
- Wash membrane with 700 µl of PE buffer.
- Centrifuge for 1 min at 16,000 x g at room temperature.
- Discard flow-through.
- Centrifuge for 1 min at 16,000 x g at room temperature.
- Discard flow-through.
- Apply 10 µl of ddH2O to the spin column (make sure the tip of the pipette is in the center of the membrane).
- Incubate for 10 min at room temperature.
- Place spin column into a clean Eppendorf tube.
- Centrifuge for 1 min at 16,000 x g at room temperature.
- Take 25 µl of the nucleosome assembly reaction and mix with 10 µl of 10x MNase buffer, 1 µl 100x BSA, 1 µl of MNase (stock at 25 U/µl) and 63 µl of water.
- Purified DNA analysis on native PAGE
2.5 µl of the purified DNA can be used for native PAGE analysis for validation purposes:- Add 2.5 µl of 2x DNA sample buffer.
- Load 5 µl of the final mix onto a 10% native PAGE (see Recipes) and run in 0.5x TBE buffer.
- Include a lane with 50 bp DNA ladder.
- Run samples at 200 V for 50 min at room temperature.
- Stain with SYBR GOLD stain solution for 10 min at room temperature.
- Image with Typhoon: Cy2/488 with PMT (photomultiplier tube) at 500 V and resolution at 100 µm.
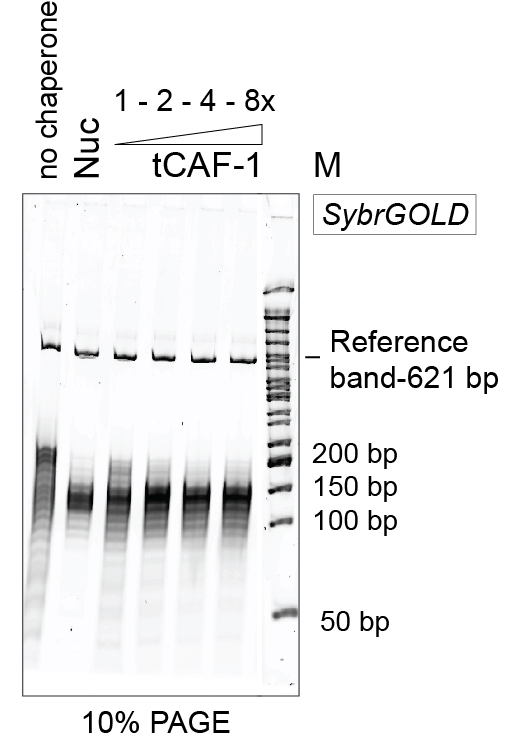
Figure 3. Example of 10% PAGE analysis of the purified DNA. Nuc stands for salt-assembled nucleosomes. tCAF-1 is an active construct of the histone chaperone CAF-1 (Mattiroli et al., 2017b). - Add 2.5 µl of 2x DNA sample buffer.
- Bioanalyzer run
Inject 1 µl of the purified DNA (at least 15-20 ng of DNA) into a DNA1000 chip in a Bioanalyzer machine.
Data analysis
The output of the Bioanalyzer run (Figure 4) is checked in the Expert 2100 Software to keep the signal threshold to 20 RFU (relative fluorescence units).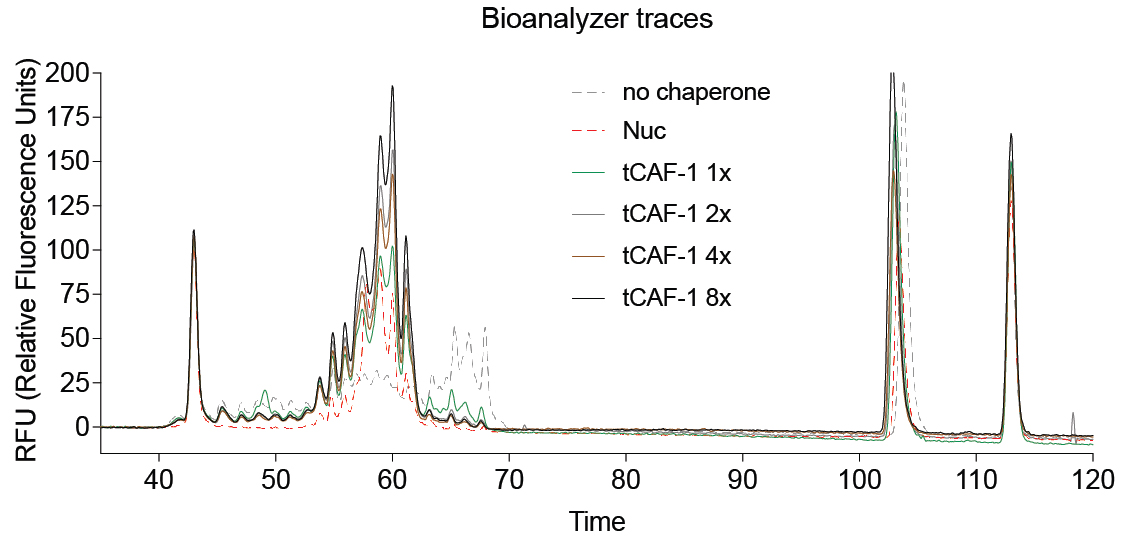
Figure 4. Example of Bioanalyzer output data. Nuc stands for salt-assembled nucleosomes. tCAF-1 is an active construct of the histone chaperone CAF-1 (Mattiroli et al., 2017b).
The data is then analyzed in Excel. We use an example to explain the analysis procedure (Figures 5 and 6).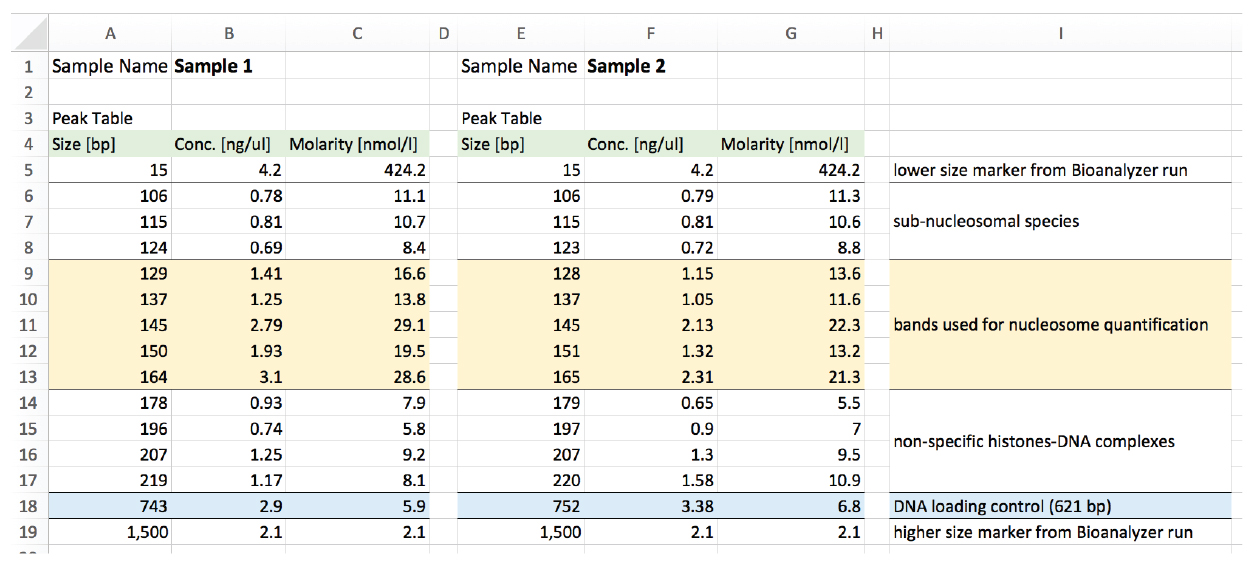
Figure 5. Example of output Bioanalyzer data of two samples. Sample 1 (columns A-C). Sample 2 (columns E-G). Column I explains the interpreted nature of the bands identified.
- Sum the molarity of the fragments with size between 126-165 bp (orange) to obtain 126-165 bp fragments [nM] (column L in Figure 6)
Sample 1: 16.6 + 13.8 + 29.1 + 19.5 + 28.6 = 107.5 nM
Sample 2: 13.6 + 11.6 + 22.3 + 13.2 + 21.3 = 82 nM - Perform loading control DNA correction to obtain loading control [nM] (column M in Figure 6)
Molarity of the loading control (C18 or G18) x Size [bp] of the loading control (A18 or E18)/621 bp (exact length of the loading control DNA)
Sample 1: 5.9 x 743/621= 7.06 nM
Sample 2: 6.8 x 752/621= 8.23 nM - Normalize the amount of 126-165 bp fragments [nM] (column L in Figure 6) to the loading control [nM] (column M in Figure 6) to obtain the Normalized values used for sample comparison (column N in Figure 6).
Sample 1: 107.5/7.06 = 15.24
Sample 2: 82/8.23 = 9.96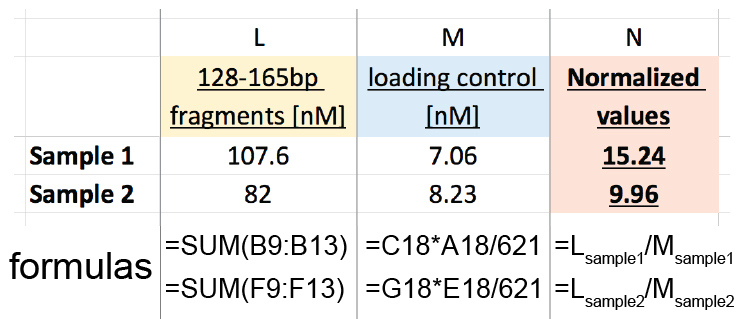
Figure 6. Analysis table example
These values are then plotted in Excel or other software (GraphPad Prism) to show the amount of nucleosome formation in each sample and to allow comparisons.
These values can be expressed as percentage of nucleosomal DNA fragments, calculated by running a known amount of untreated pure 207 bp DNA into the Bioanalyzer to deduce the conversion factor for the theoretical maximum DNA amount.
For example:
If the analysis of untreated 207 bp DNA yields a Normalized nM value of 35.
Sample 1 above will contain the following percentage (%) of nucleosome protected DNA:
% nucleosomal DNA fragmentsSample 1 = 100 x normalized valueSample 1/normalized valueuntreated DNA
% nucleosomal DNA fragmentsSample 1 = 100 x 15.24/35 = 42.8%
Notes
- Fluorescently-labeled H2A-H2B histones can be used in this assay to facilitate the identification of the nucleosome band in the assembly gel analysis.
- H2A-H2B associate with a tetrasome in absence of histone chaperones (Mattiroli et al., 2017b).
- The Bioanalyzer machine estimates the fragments size based on the elution time of the peaks and using the upper and lower marker bands as references. This leads to errors in the absolute measure of the fragment bp length. Use a salt-assembled nucleosome control to check for this variation in each experiment.
Recipes
- 1 M TCEP
0.2866 g of TCEP (Tris (2-Carboxyethyl) phosphine Hydrochloride)
360 µl of 10 M NaOH
ddH2O to 1.5 ml
Use fresh or store at -20 °C for use within one week (limit freeze/thaw cycles) - NA buffer
25 mM Tris pH 7.5 (pH measured at room temperature)
150 mM NaCl
1 mM EDTA
0.02% Tween 20
Store at 4 °C and add a final concentration of 0.5 mM TCEP just before use - SYBR Gold stain solution
Dissolve 3 µl of SYBR Gold gel stain into 50 ml of ddH2O or 0.2x TBE buffer - 10x TBE buffer (Tris/Borate/EDTA)
890 mM Tris
890 mM boric acid
20 mM EDTA pH 8.0 - 25% APS
Dissolve 0.25 g of ammonium persulfate in 1 ml of ddH2O
Store at 4 °C for not longer than 1 week - 6% PAGE (final concentrations)
6% acrylamide
0.2x TBE
0.05% APS
0.08% TEMED - 10% PAGE (final concentrations)
10% acrylamide
1x TBE
0.1% APS
0.1% TEMED - DNA sample buffer
30% (v/v) glycerol
0.25% (w/v) bromophenol blue
0.25% (w/v) xylene cyanol FF
Store at 4 °C - 3 M Na acetate pH 5.0 solution
3 M sodium acetate
Adjust pH with acetic acid to pH 5.0.
Store at room temperature
Acknowledgments
We thank Serge Bergeron for optimization of the assembly part of the protocol. F.M. is funded by EMBO (ALTF 1267-2013) and the Dutch Cancer Society (KWF 2014-6649). Research in the Luger lab is funded by the Howard Hughes Medical Institute and NIH (GM067777). The authors declare no conflicts of interest or competing interests.
References
- Dyer, P. N., Edayathumangalam, R. S., White, C. L., Bao, Y., Chakravarthy, S., Muthurajan, U. M. and Luger, K. (2004). Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol 375: 23-44.
- Elsässer, S. J. and D’Arcy, S. (2013). Towards a mechanism for histone chaperones. Biochim Biophys Acta 1819(3-4): 211-221.
- Gurard-Levin, Z. A., Quivy, J. P. and Almouzni, G. (2014). Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu Rev Biochem 83: 487-517.
- Laskey, R. A., Honda, B. M., Mills, A. D. and Finch, J. T. (1978). Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 275(5679): 416-420.
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. and Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389(6648): 251-260.
- Mattiroli, F., Gu, Y., Balsbaugh, J. L., Ahn, N. G. and Luger, K. (2017a). The Cac2 subunit is essential for productive histone binding and nucleosome assembly in CAF-1. Sci Rep 7: 46274.
- Mattiroli, F., Gu, Y., Yadav, T., Balsbaugh, J. L., Harris, M. R., Findlay, E. S., Liu, Y., Radebaugh, C. A., Stargell, L. A., Ahn, N. G., Whitehouse, I. and Luger, K. (2017b). DNA-mediated association of two histone-bound complexes of yeast Chromatin Assembly Factor-1 (CAF-1) drives tetrasome assembly in the wake of DNA replication. Elife 6.
- Muthurajan, U., Mattiroli, F., Bergeron, S., Zhou, K., Gu, Y., Chakravarthy, S., Dyer, P., Irving, T. and Luger, K. (2016). In vitro chromatin assembly: Strategies and quality control. Methods Enzymol 573: 3-41.
Article Information
Copyright
![]() Mattiroli et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Mattiroli et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Mattiroli, F., Gu, Y. and Luger, K. (2018). Measuring Nucleosome Assembly Activity in vitro with the Nucleosome Assembly and Quantification (NAQ) Assay. Bio-protocol 8(3): e2714. DOI: 10.21769/BioProtoc.2714.
- Mattiroli, F., Gu, Y., Yadav, T., Balsbaugh, J. L., Harris, M. R., Findlay, E. S., Liu, Y., Radebaugh, C. A., Stargell, L. A., Ahn, N. G., Whitehouse, I. and Luger, K. (2017). DNA-mediated association of two histone-bound complexes of yeast Chromatin Assembly Factor-1 (CAF-1) drives tetrasome assembly in the wake of DNA replication. Elife 6.
Category
Molecular Biology > Protein > Protein-DNA binding
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.