- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation and Purification of Viruses Infecting Cyanobacteria Using a Liquid Bioassay Approach


Published: Vol 8, Iss 2, Jan 20, 2018 DOI: 10.21769/BioProtoc.2691 Views: 10256
Reviewed by: Dennis NürnbergQinglu ZengAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The following protocol describes the isolation and purification of viruses infecting cyanobacteria using a liquid bioassay approach. Viruses infecting cyanobacteria are also known as cyanophages. This protocol was written specifically for the isolation of cyanophages infecting freshwater cyanobacteria particularly, cyanobacteria that cannot be cultured on solid media. The use of a clonal cyanobacterial culture is recommended for the isolation of viruses. Growth conditions (i.e., media, light cycle and temperature) should be modified based on the host of interest.
Keywords: Virus isolationBackground
Cyanobacteria are important phototrophs in both marine and freshwater systems. As other aquatic microorganisms, cyanobacteria are subject to viral infection (Suttle, 2000). For instance, in marine coastal regions, titers of viruses infecting Synechococcus spp. can reach up to 105 ml-1 and differ based on temperature, salinity and host abundance (Suttle and Chan, 1993; Waterbury and Valois, 1993). Despite the ecological importance of cyanobacteria and their viruses (also known as cyanophages), only a small number of viruses have been isolated from only a limited number of cyanobacterial strains. Consequently, it is of great interest to isolate new viruses by screening with new strains of cyanobacteria. The following protocol is relevant for both marine and freshwater systems but the example given below will focus on the isolation and purification of viruses infecting freshwater cyanobacteria (Chénard et al., 2015). The advantage of the liquid bioassay method over published protocols using solid substrate, is the opportunity to target cyanobacteria that cannot tolerate the higher temperatures often employed with the plaque assay method or cannot grow on solid media.
Materials and Reagents
- 0.45 μm or 0.22 μm syringe filters (e.g., Millex HV or GV, PVDF) (EMD Millipore)
- Sterile disposable plastic syringes, 60 ml (BD, catalog number: 309653 )
- Glass 13 x 100 mm culture tubes or 24-well plates (polystyrene, e.g., Corning, catalog number: 3524 )
- 2 ml microcentrifuge tubes
- 96-well plates (polystyrene e.g., Corning, catalog number: 3599 )
- Parafilm
- Sterile disposable plastic syringes, 5 ml (BD, catalog number: 309646 )
- Sterile hypodermic needle (18 G 1 ½, BD, catalog number: 305196 )
- Aerosol barrier filter tips (SARSTEDT, catalog number: 70.1189.215 )
- Centricon Plus-70 (30,000 NMWL, EMD Millipore, catalog number: UFC703008 )
- Slide-A-LyzerTM Dialysis cassette (20,000 MWCO, Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 66003 )
- Target host–clonal cyanobacteria strain, preferably axenic, but not necessary
- Freshwater sample (source of viruses)
- OptiprepTM density gradient medium (Axis Shield, catalog number: 1114542 )
- MilliQ water
- 0.1 N HCl
- Sodium nitrate (NaNO3)*
- Potassium phosphate dibasic (K2HPO4)*
- Magnesium sulfate heptahydrate (MgSO4·7H2O)*
- Calcium chloride dihydrate (CaCl2·2H2O)*
- Citric acid*
- Ferric ammonium citrate*
- Disodium EDTA*
- Sodium phosphate (Na2CO3)*
- Boric acid (H3BO3)*
- Manganese(II) chloride tetrahydrate (MnCl2·4H2O)*
- Zinc sulfate heptahydrate (ZnSO4·7H2O)*
- Sodium molybdate dehydrate (Na2MoO4·2H2O)*
- Copper (II) sulfate pentahydrate (CuSO4·5H2O)*
- Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O)*
- Trace metal (TM) stock (see Recipes)
- BG-11 media (see Recipes)
*Note: All chemicals used in the media recipe are reagent grade (ACS grade) and were purchased from Thermo Fisher Scientific.
Equipment
- P200 pipet (Eppendorf, catalog number: 3123000055 )
- Automatic pipet (e.g., P1000)
- Fluorometer (e.g., Turner Designs, model: TD-700 with optical kit Turner Designs, catalog number: 10-037R , excitation 350-500 nm, emission > 665 nm)
- Vortex (e.g., Scientific Industries, model: Vortex-Genie 2 )
- Ultracentrifuge (Beckman Coulter, model: Optima L-90K ) with SW-40Ti rotor (Beckman Coulter, model: SW 40 Ti , catalog number: 331302)
- Thin-walled Ultra-ClearTM open-top ultracentrifuge tubes (14 ml, 14 x 95 mm) (Beckman Coulter, catalog number: 344060 )
- Balance (micro-scale; used to balance centrifuge tubes)
- Refrigerated table top centrifuge (e.g., Eppendorf, model: 5810 R with Rotor A-4)
- Transmission Electron Microscope (TEM)
- Magnetic stir bar
- Beaker or Erlenmeyer
- Autoclave
Procedure
Definitions
Titer–concentration of infective viruses
MPN–most probable number
Lysate–a solution containing the product released from lysis of cells
- Isolation of cyanophages
- Collect ca. 50 ml water sample in a clean container (e.g., Falcon tube); pre-rinse container several times with the sample before filling. If possible, keep sample cold and in the dark until the next step.
Note: Sample volume needed will vary depending on the productivity of the environment, ranging from a few ml for productive environments, to tens of liters for oligotrophic environments. When necessary, viruses can be concentrated from large volumes of water samples to increase the detection limit. For more details on the use of ultrafiltration to isolate viruses, see Suttle et al. (1991). - Remove bacteria and plankton from the sample by filtration; use a 0.45 μm or 0.22 μm pore size syringe filter and a 60 ml plastic syringe. Store filtered sample in the dark, at 4 °C until use.
- Prepare exponentially growing target cells.
- Transfer ca. 3 ml of exponential growing cultures to 5 sterile glass 13 x 100 mm culture tubes (use a minimum of 3 tubes per water sample and 2 tubes as negative controls).
- Add 1 ml of BG-11 media (see Notes for Recipes) to 2 negative control tubes.
- Add 1 ml of the filtered water sample to each treatment tube.
- Incubate the tubes for a minimum of 7 to 10 days using the specific growth conditions of the target culture. Use a fluorometer (Turner TD700 or equivalent), to measure the in vivo fluorescence (as a proxy for growth), every 1-2 days.
- A decrease in relative fluorescence in the treatment compared with the control can be an indication of lysis by a viral agent. Clearance of the culture or distinct change in culture color can also be an indicator.
- Select tubes with suspected lytic agents.
- Repeat Steps A2 to A7, using these 4 ml samples to confirm that the lytic agent can be propagated. Use the following modifications:
- Step A4: Increase volume of cells to 4 ml.
- Steps A5 and A6: Reduce volume to 0.1 ml.
- Step A7: Expected time for lysis of culture can be shorter.
- Step A4: Increase volume of cells to 4 ml.
- If lysis is observed, aliquot the lysate into triplicate with a 0.45 μm or 0.22 μm pore size filter to remove most of the bacteria.
- Collect ca. 50 ml water sample in a clean container (e.g., Falcon tube); pre-rinse container several times with the sample before filling. If possible, keep sample cold and in the dark until the next step.
- Obtaining pure cyanophage stock
- Determine the titer of the lysate using the 96-well plate MPN assay (Suttle and Chan, 1993)
- Prepare exponentially growing target cells. Add fresh BG-11 media to the exponentially growing culture (1:1). Dispense 150 μl of diluted cells to each well.
- Set up a 10-fold dilution series of the lysate. For example, add 1,350 μl of BG-11 media to 8 x 2 ml microcentrifuge tubes. Add 150 μl of lysate to first tube (1/10 dilution). Mix by vortexing. Repeat until the last dilution (1/109 dilution).
- Add one dilution per column by adding 50 μl of the proper dilution to each well. Each column consists of 8 wells. One to two columns of the 96-well plate is used as a control which means 50 μl of BG-11 media is added to each of the wells (e.g., columns 1 and 12, Figure 1).
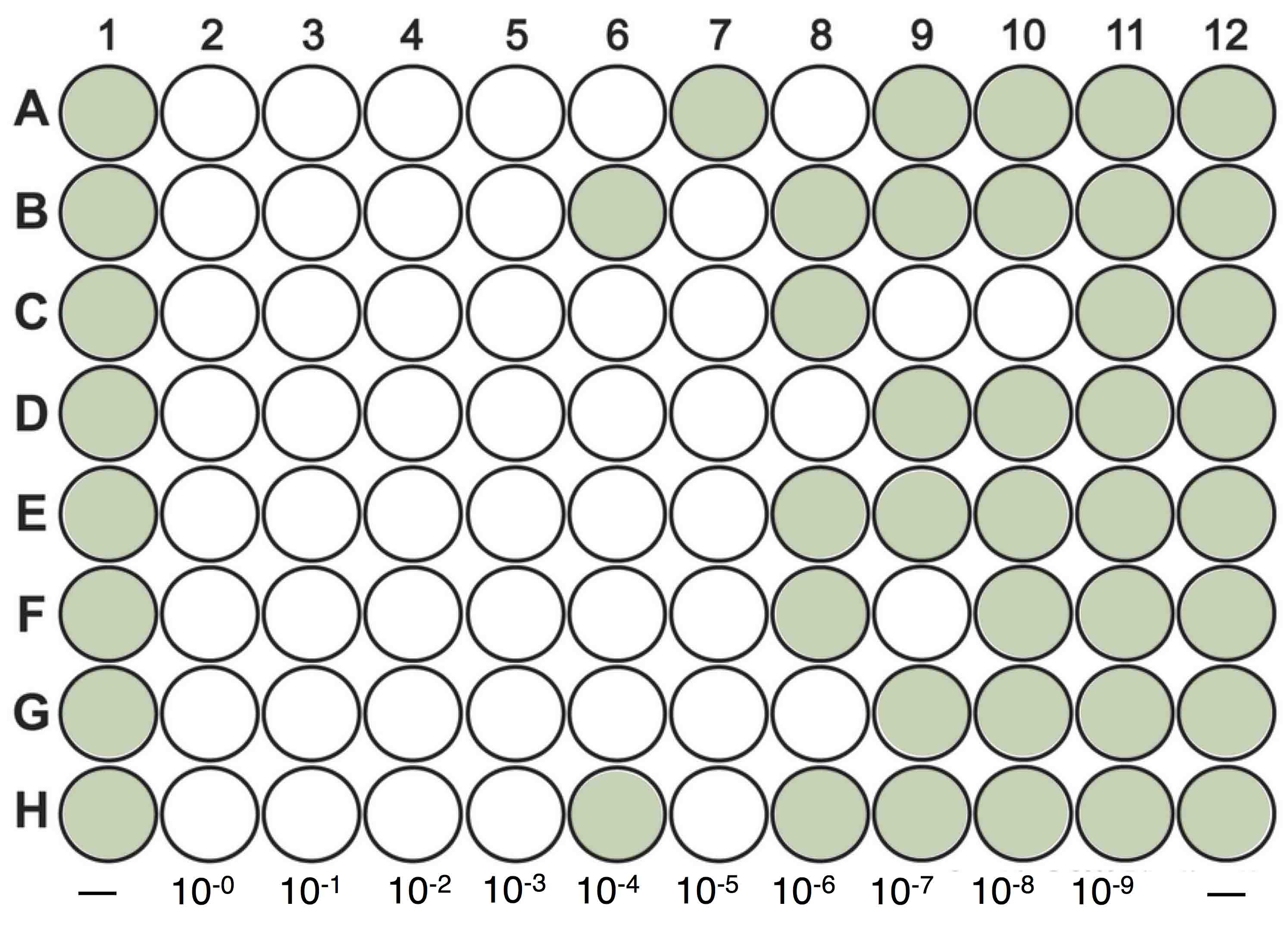
Figure 1. Diagram showing a 96-wells plate MPN assay. Columns 1 and 12 represent control cultures, where 50 μl of BG-11 media is added into each of the wells. 50 μl of a 10-fold dilution series (10-0 to 10-9) of the lysate is added to the remaining columns. The plate is then monitored after 7 days to record the number and position of clear wells. - Seal the 96-well plates with Parafilm, and incubate the plates using the specific conditions of the cyanobacteria cultures.
- Monitor plates for lysis every few days, recording the number and position of clear wells on the plate. When new clear or nearly clear wells no longer appear for 1 week, record the final ‘score’ for each dilution level. (Figure 1)
- Determine the concentration of infective virus, using a Most Probable Number (MPN) calculation program* (Jarvis et al., 2010).
*Note: The program can be downloaded from: http://www.wiwiss.fu-berlin.de/fachbereich/vwl/iso/ehemalige/wilrich/index.html.
- Prepare exponentially growing target cells. Add fresh BG-11 media to the exponentially growing culture (1:1). Dispense 150 μl of diluted cells to each well.
- Purification of a clonal virus isolate (see Middelboe et al., 2010 In: Manual of Aquatic Viral Ecology. The American Society of Limnology and Oceanography p118-133 for more details)
- Prepare exponentially growing target cells, dispense ca. 4 ml into 13 x 100 mm culture tubes.
- Use BG-11 media to dilute lysate of known concentration to 1 infective virus/ml.
- Add 0.2 ml (0.2 infectious units) to each of 20 tubes of susceptible host cells.
- Monitor tubes (by eye or by in vivo fluorescence) for 2 to 3 weeks. Cultures in which lysis occurs are assumed to be the result of a single-virus infection.
Notes: - If lysis occurs in ≤ 4 tubes out of 20, it is assumed that lysis in each tube was caused by one infectious unit, therefore each tube would contain a separate phage clone. Transfer a small aliquot from all the tubes to fresh target cells to confirm the results. Proceed to Step B2e.
- If lysis occurs in more than 4 tubes, repeat Steps B2a-B2d, but reduce the volume of diluted lysate added in Step B2c accordingly in order to achieve lysis in ≤ 4 tubes out of 20 (e.g., add 0.1 ml instead of 0.2 ml).
- Scale up each lysed culture (clonal isolate) to make primary phage stocks; e.g., add 5 μl of 0.45 μm filtered lysate to 40 ml of exponentially growing cells.
- Incubate the tubes for 7 to 10 days using the specific growth conditions of the culture. Once the culture has lysed, centrifuge the lysate (5,000 x g, 15 min) to remove most of the cellular debris. Carefully transfer the supernatant to a fresh tube. Afterwards, filter the supernatant with a 0.22 μm pore size syringe filter (PVDF, Millipore), and store at 4 °C in the dark until further use. This is the primary phage stock solution.
- The phage stock can be further concentrated using a centrifugal UF device such as the Centricon Plus-70 (follow the manufacturer’s instructions).
- Pre-rinse the Centricon Plus-70 with 50 ml of MilliQ water.
- Centrifuge at 3,500 x g for 10 min. Pour out the filtrate.
- Add 40 ml of sample into the centricon.
- Centrifuge at 3,500 x g for 15 min. To collect the viral concentrate, connect the Centricon to the concentration collection cup, invert the Centricon, and remove the filtrate collection cup. Centrifuge at 1,000 x g for 2 min. The final volume should be less than 1 ml. Store concentrate at 4 °C until use.
- Prepare exponentially growing target cells, dispense ca. 4 ml into 13 x 100 mm culture tubes.
- Determine the titer of the lysate using the 96-well plate MPN assay (Suttle and Chan, 1993)
- Purification of the cyanophage stock using an OptiprepTM gradient and the SW40 rotor (see Lawrence and Steward, 2010 for more details)
Note: The viral concentrate is further purified on a continuous, isopycnic density gradient.- Prepare OptiprepTM solutions using culture media as the diluent. We used 20%, 30% and 40% (v/v) final-OptiprepTM concentration solutions.
- Using a syringe and needle, prepare 3-step gradients with thin-walled open-top ultracentrifuge tubes (e.g., Beckman Ultra-ClearTM tube, 14 ml, 14 x 95 mm), beginning with the least dense solution. Be sure to leave enough room at the top of the centrifuge tube to layer the sample.
- Allow the gradient to equilibrate for at least 2 h at room temperature (or overnight at 4 °C).
- Carefully overlay ~0.2 to 1 ml of the viral concentrate on top of the OptiprepTM gradient using an adjustable pipet fitted with an aerosol barrier tip. Balance the underweight tubes by adding culture media one drop at a time, being careful not to disturb the gradient (use a fine-tipped transfer pipet).
- Carefully load the tubes into rotor and ultracentrifuge. For a 12 ml gradient with 1 ml virus concentrate sample in a Beckman Coulter SW-40Ti, centrifuge for 8 h at 207,570 x g (34,100 rpm) and 20 °C.
- Once the run is finished, carefully remove the tube from the rotor and clamp it vertically on a stand. Look for the presence of band(s) in the OptiprepTM gradient and remove the band(s) by side puncturing the tube with a needle. In a darkened room, shine a light from the top of the tube to help to visualize the bands. Gentle extract the band with the syringe and transfer the contents to a small tube (see Lawrence and Stewart, 2010 for more details).
Note: OptiprepTM should not affect the infectivity of the viral isolates, however, proper controls should be performed in order to eliminate any possible negative effects that OptiprepTM may have on the growth of specific viral-hosts. - To remove any leftover OptiprepTM material from the sample, a Slide-A-LyzerTM Dialysis cassette can be used. Once the band sample is loaded into the cassette according to the manufacturer’s instructions, the cassette is stored overnight at 4 °C into a beaker of fresh and sterile BG-11 media. Afterward, the dialysis sample is removed from the dialysis cassette according to the manufacturer’s instructions.
- Prepare OptiprepTM solutions using culture media as the diluent. We used 20%, 30% and 40% (v/v) final-OptiprepTM concentration solutions.
- Confirmation of virus isolate
- To confirm that the lytic agent of the cyanobacteria was purified, a subsample of the purified band (from Step C7), is diluted and added back to an exponentially growing culture of cyanobacteria.
- Once the culture is lysed, filter the lysate through a 0.45 μm or 0.22 μm pore size filter (PVDF), titer (as in Step B1) and store the stock at 4 °C in the dark.
- A subsample of the purified virus can also be used for examination on a Transmission Electron Microscope (TEM) to confirm that a virus is the lytic agent and to examine its morphology.
Samples might need to be concentrated using the centricon or by ultracentrifugation. In general, a minimum of 1010 virus particles/ml are need for TEM examination using the negative staining procedure. More details on the TEM examination can be found in Suttle and Chan, 1993.
- To confirm that the lytic agent of the cyanobacteria was purified, a subsample of the purified band (from Step C7), is diluted and added back to an exponentially growing culture of cyanobacteria.
Recipes
Notes:
- Stock solutions can be conveniently prepared at 1,000x for each ingredient. Add 1 ml of each 1,000x stock to prepare 1 L of media. Store stock solutions at 4 °C.
- When preparing the trace metal stock mix, dissolve each ingredient separately prior to adding the next one from the list.
- Trace metal (TM) stock
2.86 g/L H3BO3
10 mg/L CuSO4·5H2O
10 mg/L Co(NO3)2·6H2O
6 mg/L Na2MoO4·2H2O
22 mg/L ZnSO4·7H2O
180 mg/L MnCl2·4H2O - BG-11 media
Notes:- A freshwater media for the culture of cyanobacteria; ref Ripka et al. (1979), modified as described below
- We use the trace metal stock (TM) recipe for F/2 media, more as a matter of convenience. The composition is very similar to the TM stock for BG-11.
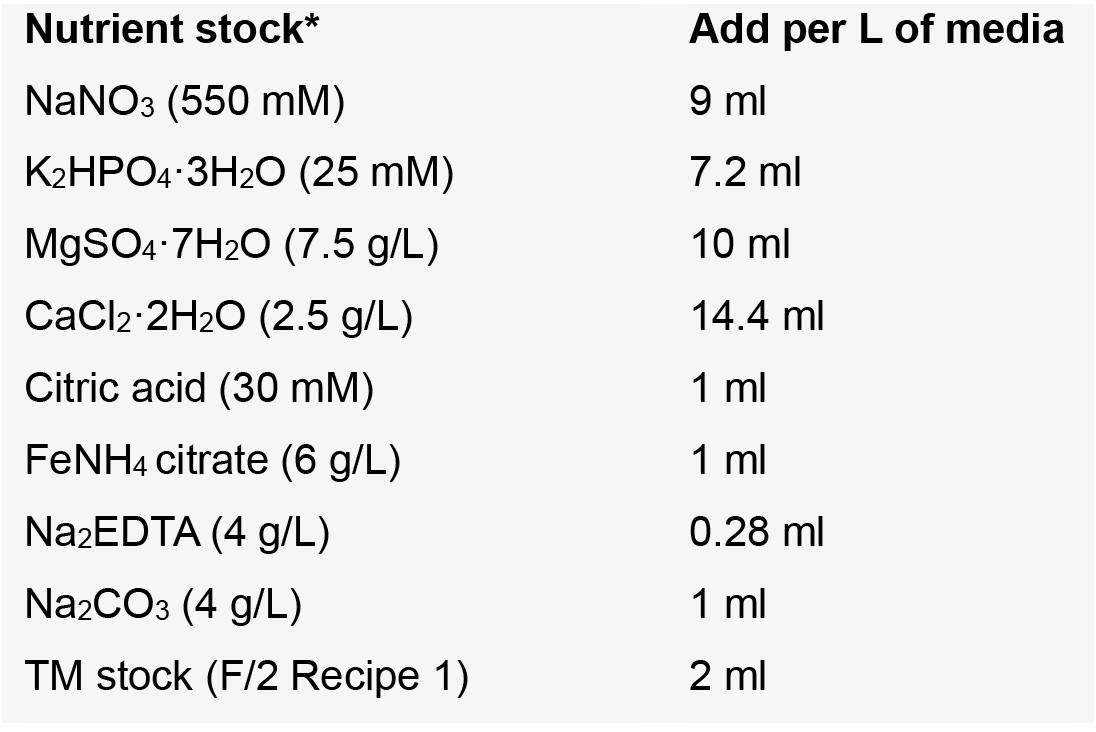
- To prepare 1 L of media, carefully slide a clean magnetic stir bar into a clean acid-washed beaker or Erlenmeyer. Add 1 L of Milli-Q water, and place the container onto a magnetic stir plate
- With gentle stirring, add the above nutrients in the order specified using either an automatic pipet (e.g., P1000) fitted with sterile tips or sterile serological pipets
- Adjust pH to ca. 8.0 (initial pH is ca. 8.5) using 0.1 N HCl (a couple of drops)
- Once thoroughly mixed, dispense the media into 500 ml portions and autoclave for 20 min at 121 °C
- A freshwater media for the culture of cyanobacteria; ref Ripka et al. (1979), modified as described below
Acknowledgments
CC is supported by the Singapore’s National Research Foundation under its Marine Science Research and Development Programme (Award No. MSRDP-P13). Development of these protocols is supported in part by the Canadian Foundation for Innovation, Leaders of Opportunity Fund (Project # 25412) and National Sciences and Engineering Research Council of Canada (EQPEQ 375995-09). This protocol was inspired and adapted from previous works: Middelboe et al. (2010); Suttle and Chan (1993); Chénard et al. (2015). The authors do not have any conflicts of interest or competing interests to declare.
References
- Chénard, C., Chan, A. M., Vincent, W. F. and Suttle, C. A. (2015). Polar freshwater cyanophage S-EIV1 represents a new widespread evolutionary lineage of phages. ISME J 9(9): 2046-2058.
- Jarvis, B., Wilrich, C. and Wilrich, P. T. (2010). Reconsideration of the derivation of Most Probable Numbers, their standard deviations, confidence bounds and rarity values. J Appl Microbiol 109(5): 1660-1667.
- Lawrence, J. E. and Steward, G. F. (2010). Purification of viruses by centrifugation. In: Wilhelm, S. W., Weinbauer, M. G. and Suttle, C. A. [Eds.]. Manual of Aquatic Viral Ecology. ASLO pp: 116-181.
- Middelboe, M., Chan, A. M., and Bertelsen, S. K. (2010). Isolation and life cycle characterization of lytic viruses infecting heterotrophic bacteria and cyanobacteria. In: Wilhelm, S. W., Weinbauer, M. G. and Suttle, C. A. [Eds.]. Manual of Aquatic Viral Ecology. ASLO pp: 118-133.
- Ripka, R., Deruelles, J., Waterbury, J. B., Herdman, M. and Stainer, R. Y. (1979). Generic assignment, strain histories and properties of pure cultures of cyanobacteria. J Gen Microbiol 111: 1-61.
- Suttle, C. A. (2000). The ecological, evolutionary and geochemical consequences of viral infection of cyanobacteria and eukaryotic algae. In: Hurst, C. J. (Ed.). Chapter 6 (248-296) in Viral Ecology. Academic Press pp: 639.
- Suttle C. A. and Chan, A. M. (1993). Marine cyanophages infecting oceanic and coastal strains of Synechococcus: abundance, morphology, cross-infectivity and growth characteristics. Mar Ecol Prog Ser 92: 99-109.
- Suttle C. A., Chan, A. M. and Cottrell, M. T. (1991). Use of ultrafiltration to isolate viruses from seawater which are pathogens of marine phytoplankton. Appl Environ Microbiol 57 (3): 721-726.
- Waterbury, J. B. and Valois, F. W. (1993). Resistance to co-occuring phages enables marine Synechococcus communities to coexist with cyanophages abundant in seawater. Appl Environ Microbiol 59: 3393-3399.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chénard, C. and Chan, A. M. (2018). Isolation and Purification of Viruses Infecting Cyanobacteria Using a Liquid Bioassay Approach. Bio-protocol 8(2): e2691. DOI: 10.21769/BioProtoc.2691.
Category
Microbiology > Microbial cell biology > Cell isolation and culture
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.