- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Method for Multiplexing CRISPR/Cas9 in Saccharomyces cerevisiae Using Artificial Target DNA Sequences

Published: Vol 7, Iss 18, Sep 20, 2017 DOI: 10.21769/BioProtoc.2557 Views: 13368
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Genome manipulation has become more accessible given the advent of the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) editing technology. The Cas9 endonuclease binds a single stranded (single guide) RNA (sgRNA) fragment that recruits the complex to a corresponding genomic target sequence where it induces a double stranded break. Eukaryotic repair systems allow for the introduction of exogenous DNA, repair of existing mutations, or deletion of endogenous gene products. Targeting of Cas9 to multiple genomic positions (termed ‘multiplexing’) is achieved by the expression of multiple sgRNAs within the same nucleus. However, an ongoing concern of the CRISPR field has been the accidental targeting of Cas9 to alternative (‘off-target’) DNA locations within a genome. We describe the use (dubbed Multiplexing of Cas9 at Artificial Loci) of installed artificial Cas9 target sequences into the yeast genome that allow for (i) multiplexing with a single sgRNA; (ii) a reduction/elimination in possible off-target effects, and (iii) precise control of the placement of the intended target sequence(s).
Keywords: CRISPR/Cas9Background
The CRISPR (Clustered Regularly Interspaced Palindromic Repeats) mechanism has evolved in prokaryotes as a primitive adaptive immune system with the capability to edit any genome with great precision (Jinek et al., 2012; Sorek et al., 2013). This biotechnology requires the use of an endonuclease (Cas9) from S. pyogenes (or othologous species), a single RNA ‘guide’ sequence, and exogenous donor DNA (if needed). In only a few years, CRISPR/Cas9 has been utilized in numerous research laboratories in both academic and industry settings to target DNA sequences within any genome (Doudna and Charpentier, 2014). A variety of research areas including basic research, biofuels, agriculture, genetic disorders, and human pathogens/disease have begun harnessing this technology to address important scientific questions (Estrela and Cate, 2016; Demirci et al., 2017; Men et al., 2017). Recent work in S. cerevisiae has piloted the development of novel CRISPR-based applications including automated genomic engineering (Si et al., 2017), chromosome splitting (Sasano et al., 2016), and the use of nuclease-dead Cas9 (dCas9) to modulate gene expression (Jensen et al., 2017). While this editing system has proved extremely useful, a number of concerns are still being actively addressed. These include off-target effects–the propensity of Cas9 to accidentally target additional genomic positions (Cho et al., 2014; Zhang et al., 2015), the required cloning step(s) needed to generate multiple sgRNAs for Cas9 multiplexing (Ryan and Cate, 2014), and the safety and application of Cas9-based ‘gene drives’ (DiCarlo et al., 2015). Our methodology addresses some of these issues by engineering artificial Cas9 target site(s) within the yeast genome. We describe (i) the selection of the artificial sequences used to multiplex Cas9; (ii) the cloning strategies used to construct plasmids harboring the unique target sites flanking several genes including Cas9 itself; (iii) integration of these constructs into a single yeast genome in successive steps, and (iv) editing using expressed Cas9, sgRNA, and donor DNA to demonstrate proof of concept. This system allows for seamless, marker-less, multi-loci genomic editing with only a single sgRNA. We envision this method could be useful for synthetic genome construction, yeast library generation, and simultaneous manipulation of related genes within a common genetic or signaling pathway.
Materials and Reagents
- Pipette tips (LTS tips 1,000 μl, 250 μl, 20 μl, Mettler-Toledo, Rainin, catalog numbers: GPS-L1000 , GPS-L250 , and GPS-L10 )
- Tubes (Axygen Microtubes 1.5 ml clear, homo-polymer, boil-proof) (Corning, Axygen®, catalog number: MCT-150-C )
- Disposable sterile plastic 15 ml (Corning, Falcon®, catalog number: 352099 ) and 50 ml (Corning, Falcon®, catalog number: 352098 ) conical centrifuge tubes
- Disposable glass test tubes (20 x 150 mm) (Fisher Scientific, catalog number: 14-958K )
- Kimble Kim-Kap test tube closures (Fisher Scientific, catalog number: 14-957-91C)
Manufacturer: DWK Life Sciences, Kimble®, catalog number: 7366020 . - Plastic Petri dish (100 x 15 mm size) (VWR, catalog number: 25384-088 )
- 0.5 mm glass beads (Bio Spec Products, catalog number: 11079105 )
- Yeast strains
- SF838-1Dα (MATα ura3-52 leu2-3,122 his4-519 ade6 pep4-3 gal2) (Univ. of Oregon; Rothman and Stevens, 1986)
- THS4218 (SF838-1Dα; HIS4 his3Δ::HygR) used for in vivo plasmid assembly and recovery (Univ. of California, Berkeley; Finnigan and Thorner, 2015)
- BY4741 (MATα his3∆1 ura3∆0 leu2∆0 met15∆0) (Univ. of California, Berkeley; Brachmann et al., 1998) for construction of all yeast strains tested
- SF838-1Dα (MATα ura3-52 leu2-3,122 his4-519 ade6 pep4-3 gal2) (Univ. of Oregon; Rothman and Stevens, 1986)
- One Shot® TOP10 chemically competent E. coli (Thermo Fisher Scientific, InvitrogenTM, catalog number: C404003 )
Note: See (Finnigan and Thorner, 2015) for propagation and preparation of TOP10 seed cultures. - Plasmid containing prGalL-Cas9-CYC1(t) used as template DNA for construction of Cas9-containing cassettes (Addgene, catalog number: 43804) from (DiCarlo et al., 2013)
- Plasmid containing prSNR52-sgRNA-SUP4(t) (Addgene, catalog number: 43803 ; synthesized de novo by Genscript) (DiCarlo et al., 2013)
- ssDNA: deoxyribonucleic acid sodium salt from salmon testes (boiled for 10 min and cooled on ice prior to each use of a 10 mg/ml stock solution in water) (Sigma-Aldrich, catalog number: D1626 )
- Zymolyase® 100T from Arthrobacter leuteus (25 mg/ml) (Amsbio, catalog number: 120493-1 ) in 50% glycerol stock (certified ACS grade) (Fisher Scientific, catalog number: G33 )
- Appropriate restriction enzyme(s) for digestion
- QIAquick gel extraction kit (QIAGEN, catalog number: 28706 )
- T4 DNA ligase (New England Biolabs, catalog number: M0202 )
- GeneJET plasmid miniprep kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K0503 )
- KOD hot start DNA polymerase (EMD Millipore, catalog number: 71086-3 , distributed by VWR, catalog number: 80511-384)
- Custom DNA oligonucleotide primers (25-100 nmol concentration; Integrated DNA Technologies)
- GeneJet PCR purification kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K0701 )
- Ethidium bromide (Sigma-Aldrich, catalog number: E8751 )
- Agarose powder (U.S. Biotech Sources, catalog number: G01PD-500 )
- Hygromycin, used at 300 μg/ml (Thermo Fisher Scientific, GibcoTM, catalog number: 10687010 )
- G418 sulfate (Geneticin), used at 200 μg/ml (Thermo Fisher Scientific, GibcoTM, catalog number: 11811031 )
- D-(+)-Raffinose pentahydrate (20% stock in water, filter sterilized, not autoclaved) (Sigma-Aldrich, catalog number: R7630 ). Filtered using disposable cellulose nitrate filter (0.2 μm filter size) (Corning, catalog number: 430186 )
- Sucrose (20% stock in water, filter sterilized, not autoclaved) (Fisher Scientific, catalog number: S3 )
- 1 M lithium acetate dihydrate (CH3COOLi·2H2O, reagent grade) (Sigma-Aldrich, catalog number: L6883 )
- 50% PEG: Poly (ethylene glycol), BioXtra avg. molecular weight 3,350 (Sigma-Aldrich, catalog number: P4338 )
- Ampicillin (final concentration of 100 μg/ml) (RPI, catalog number: A40040-100.0 )
- Kanamycin (final concentration of 50 μg/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 11815024 )
- Yeast extract (BD, BactoTM, catalog number: 212750 )
- Peptone (BD, BactoTM, catalog number: 211677 )
- Dextrose (20% stock in water) (Thermo Fisher Scientific, catalog number: D16 )
- D-(+)-Galactose (20% stock in water, filter sterilized, not autoclaved) (Sigma-Aldrich, catalog number: G0750 )
- Tryptone (BD, BactoTM, catalog number: 211705 )
- Sodium chloride (NaCl, certified ACS grade ≥ 99.0%) (Fisher Scientific, catalog number: S271 )
- Potassium chloride (KCl, BioXtra ≥ 99.0%) (Sigma-Aldrich, catalog number: P9333 )
- Magnesium chloride hexahydrate (MgCl2·6H2O, BioXtra ≥ 99.0%) (Sigma-Aldrich, catalog number: M2670 )
- Magnesium sulfate solution (MgSO4, molecular biology grade) (Sigma-Aldrich, catalog number: M3409 )
- SuperPure agar, bacteriological grade (US Biotech Sources, catalog number: A01PD-500 )
- Yeast nitrogen base minus amino acids and minus ammonium sulfate (Sigma-Aldrich, catalog number: Y1251 )
- Ammonium sulfate ((NH4)2SO4 certified ACS grade ≥ 99.0%) (Fisher Scientific, catalog number: A702 )
- ‘Almost complete’ amino acid mixture
Adenine HCl (Sigma-Aldrich, catalog number: A9795 ), 20 mg/L
Arginine (Sigma-Aldrich, catalog number: A5131 ), 20 mg/L
Tyrosine (Sigma-Aldrich, catalog number: T3754 ), 30 mg/L
Isoleucine (Sigma-Aldrich, catalog number: I2752 ), 30 mg/L
Phenylalanine (Sigma-Aldrich, catalog number: P2126 ), 50 mg/L
Glutamic acid (Sigma-Aldrich, catalog number: G1251 ), 100 mg/L
Aspartic acid (Sigma-Aldrich, catalog number: A9256 ), 100 mg/L
Threonine (Sigma-Aldrich, catalog number: T8625 ), 200 mg/L
Serine (Sigma-Aldrich, catalog number: S4500 ), 400 mg/L
Valine (Sigma-Aldrich, catalog number: V0500 ), 150 mg/L - Methionine (Sigma-Aldrich, catalog number: M9625 )
- Lysine (Sigma-Aldrich, catalog number: L5626 )
- Histidine (Sigma-Aldrich, catalog number: H8125 )
- Leucine (Sigma-Aldrich, catalog number: L8000 )
- Uracil (Sigma-Aldrich, catalog number: U0750 )
- 5-Fluoroorotic acid (5-FOA) (Oakwood Products, catalog number: 003234 )
- Sodium hydroxide (NaOH certified ACS grade ≥ 97.0%) (Fisher Scientific, catalog number: S318 )
- Tris base, molecular biology grade ≥ 99.8% (Fisher Scientific, catalog number: BP152 )
- Glacial acetic acid (certified ACS grade) (Fisher Scientific, catalog number: A38 )
- Ethylenediaminetetraacetic acid (EDTA 99%-101%) (Fisher Scientific, catalog number: S311 )
- Ultrapure sterile water (Millipore Sigma, Milli-Q water purification system)
- YPD liquid media (see Recipes)
- YPGal (see Recipes)
- SOC medium (see Recipes)
- YPD plates (with appropriate drugs optional) (see Recipes)
- Synthetic drop-out plates/media (see Recipes)
- 5-FOA plates (see Recipes)
- LB plates (with appropriate drug included) (see Recipes)
- TAE buffer (see Recipes)
Equipment
- Pipettes (Rainin, Pipet-Lite LTS, 1,000 μl, 250 μl, 20 μl, and 2 μl sizes)
- PCR machine (MJ Research PTC-200 Peltier Thermo Cycler, dual 30-well alpha blocks) (MJ Research, model: PTC-200 )
- Centrifuge (Eppendorf microcentrifuge) (Eppendorf, model: 5415 D , catalog number: 022621408)
- Eppendorf rotor (for 24 x 1.5/2 ml) (Eppendorf, model: F-45-24-11 , catalog number: 022636502)
- Vortexing adaptor (Microtube foam insert for Fisher Vortex Genie 2 mixer) (Scientific Industries, model: Vortex Genie 2 , catalog number: 504-0234-00)
- Ice maker (Hoshizaki American)
- Water bath (Thermomix circulating water bath, Model B, type 852 013/5)
- DNA gel electrophoresis apparatus (HE 33 Mini Submarine Unit) (GE Healthcare, catalog number: 80-6052-45 )
- ChemiDoc UV transilluminator (Bio-Rad Laboratories, model: ChemiDocTM XRS+, catalog number: 1708265 )
- Incubator rotator (Labquake shaker) (Labindustries, model: T-415-110 )
- Incubators (VWR, model: Model 1535 )
- Autoclave (Univ. of California, Barker Hall)
Procedure
Here we describe the strategy used to design, construct, integrate, and edit with Cas9 artificial target sequences within the yeast genome. We have focused heavily on the cloning methodologies for how to precisely install the programmed target sites (other methods may be substituted at certain steps and we highlight these) and multiplex with Cas9 in vivo. We provide a detailed description of our published (Finnigan and Thorner, 2016) proof of concept describing this methodology using both an essential and non-essential gene as well as the Cas9 gene itself to illustrate the utility and power of this approach.
- Selection of artificial site(s)
- To begin, a unique target sequence (designated ‘[u1]’) of a minimum of 23 bp (20 target + 3 Protospacer Adjacent Motif [PAM]) must be selected that has a maximum mismatch to the organism of choice (budding yeast genome) and all other exogenous components of the system (added vectors, GFP, drug cassettes, tags, etc.). The chosen sequence may be from another organism (in our case, two human gene sequences were selected), or may be generated de novo.
Note: Other studies have found a variety of limitations to the target sequence (and PAM). It is recommended (in yeast) to not have a poly-T sequence (five or more) as this represents a termination signal for RNA Pol III (DiCarlo et al., 2013). Additionally, sequences that are extremely AT or GC rich are not recommended. - Putative artificial 23 bp sequences should be checked against the yeast genome (and other exogenous sequences) using the NCBI Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). An initial screening of the 3’ most 15 bp (including the PAM) should first be searched. This represents the ‘seed’ region used by the Cas9/sgRNA complex to first associate with a putative matching genomic target sequence (Jinek et al., 2012; DiCarlo et al., 2013; Jiang et al., 2013). A maximum mismatch between these bases should be identified with particular emphasis on mismatches within the GG bases of the PAM sequence. Once putative target(s) are found, a full search of the entire 23 bp sequence can be interrogated against the yeast genome with the same preference for a high level of mismatch.
Note: If the artificial target is to be included within the open reading frame (and translated as an appended N- and/or C-terminal tag), then an extra base (23 + 1) should be included to keep the sequence in-frame. Moreover, the occurrence of any in-frame stop codon within the sequence should be avoided. - Design of this multiplexing system using a minimum number of sgRNAs should be carefully considered prior to the construction of any assembled plasmids and/or genome integration. For instance, the total number of unique site(s) and their placement across gene(s) may be difficult to undo once the initial investment of strain construction has been made. Our system utilizes a [u1] target site flanking multiple loci to be manipulated whereas the Cas9 gene itself is flanked by either the [u1] or [u2] sites providing the option for either simultaneous or sequential seamless excision of the nuclease.
- To begin, a unique target sequence (designated ‘[u1]’) of a minimum of 23 bp (20 target + 3 Protospacer Adjacent Motif [PAM]) must be selected that has a maximum mismatch to the organism of choice (budding yeast genome) and all other exogenous components of the system (added vectors, GFP, drug cassettes, tags, etc.). The chosen sequence may be from another organism (in our case, two human gene sequences were selected), or may be generated de novo.
- Construction of tagged genes, Cas9, and sgRNA plasmids
- Following selection of the unique [u1, u2, u3…] artificial sites (Procedure A), as well as the intended strategy to flanking the loci of interest with said sites (Figure 1, bottom), in vivo plasmid assembly (Finnigan and Thorner, 2015) can be used to install the sites at their precise position(s).
Note: We have previously described the entire process for plasmid ‘in vivo ligation’ followed by recovery of the constructed vectors from yeast (Finnigan and Thorner, 2015). We will include additional comments/steps pertaining to pertinent modifications from the original protocol.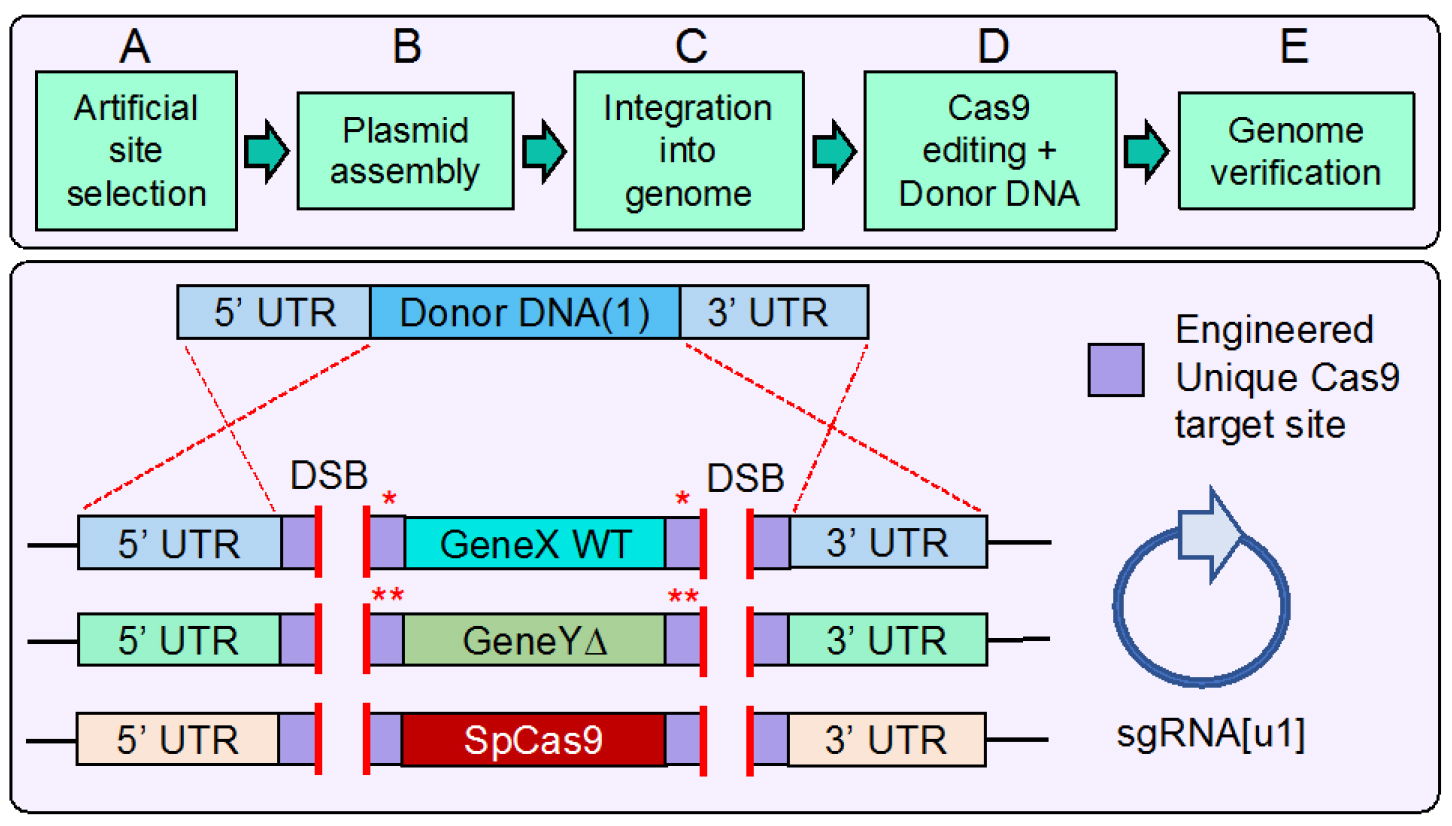
Figure 1. Design, construction, and use of artificial DNA sequences for Cas9 multiplex gene editing in budding yeast. Top. Workflow overview. This protocol begins with selection of the artificial DNA Cas9 target sites followed by construction of the desired plasmid(s) including Cas9 and sgRNA expression cassettes. Next, successive integration events into the yeast genome place the target sites at multiple loci including Cas9. Transformation of the sgRNA plasmid and amplified donor DNA fragment(s) in the presence of Cas9 allows for in vivo editing. Finally, verification of the genome by diagnostic PCR and DNA sequencing confirms the intended modifications. Bottom. An example genome containing six identical [u1] (unique engineered sequence) sites at three loci (one of which includes Cas9). The 24 bp sequence (20 target + 3 PAM + 1 extra base) is appended as an 8-residue tag at both the N- and C-termini of Cdc11 (single red asterisk). In the case of the deleted GeneY (Shs1), the added [u1] sites (23 bp) are part of the UTR (two red asterisks). Use of only one sgRNA allows for double stranded break (DSB) formation at all six genomic positions. The presence of corresponding amplified donor DNA (only one is illustrated for clarity) with flanking homology to the UTR allows for simultaneous multi-gene marker-less integration events via homologous recombination. Depending on the identity of the artificial site [u1 or u2] flanking the Cas9 gene, concomitant or sequential excision of Cas9 can be achieved depending on the need for future editing events in the given strain. - Design of oligonucleotides should include the intended 23 (or 24) bp artificial sequence and place either in-frame as part of the gene to be tagged, or within the flanking UTR by a two-step PCR amplification. As shown in Figure 2, we describe the construction process for two identical [u1] tags at the N- and C-termini of a given gene. However, we have chosen to separate inclusion of each tag into two intermediate plasmids to be built in parallel. If a single plasmid assembly process was used, then there would be competition between PCR fragments containing the intended [u1] sequences; it is possible that rather than include the WT gene between the two artificial sites, exclusion of the central fragment could result and create only a single [u1] site. To eliminate this possibility, we constructed each [u1] sequence separately. This may be useful in other applications where a gene might purposely be flanked by two different artificial sites.
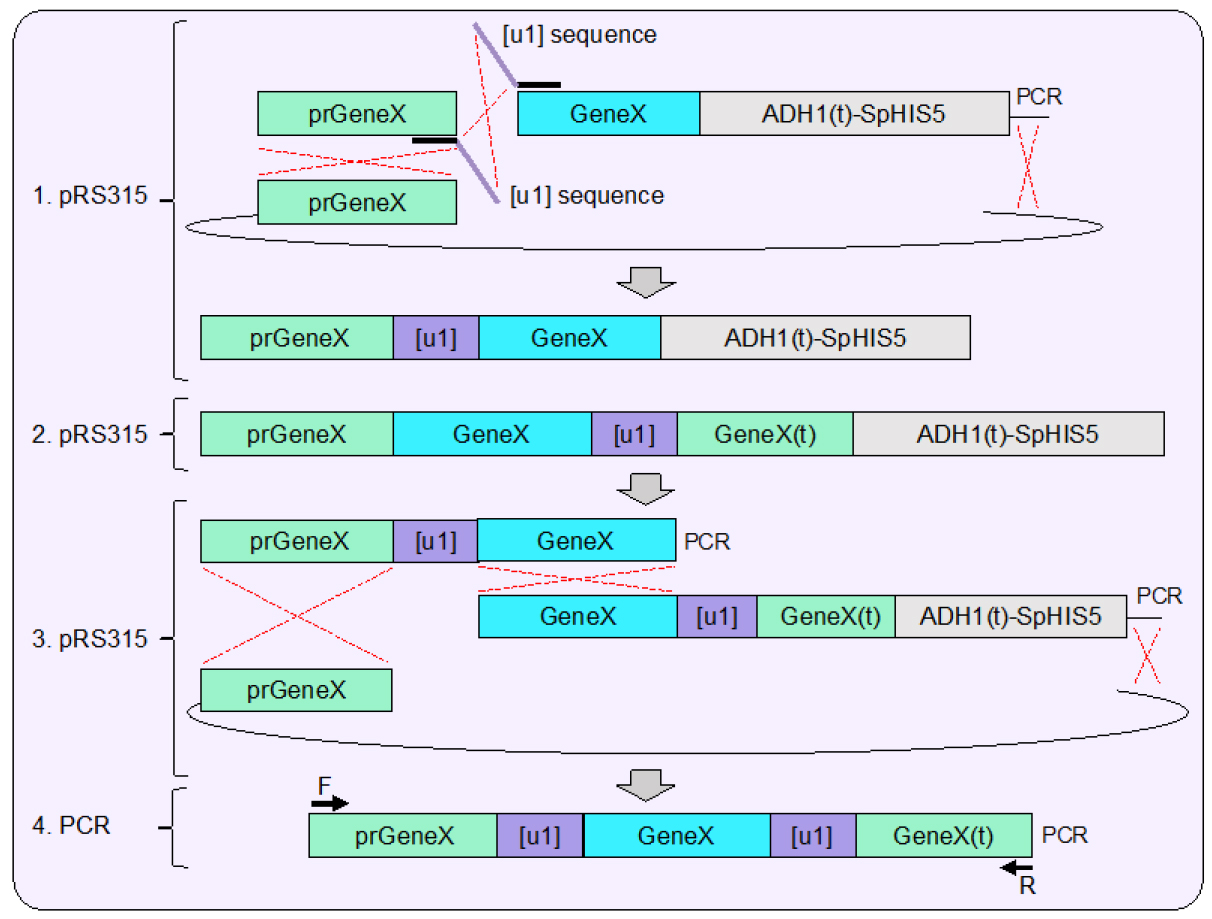
Figure 2. Plasmid assembly of sample gene containing flanking unique sequence sites [u1]. 1. Using in vivo plasmid assembly in yeast (Finnigan and Thorner, 2015) one artificial site [u1] is inserted between the generic promoter (prGeneX) and open reading frame of a sample gene (GeneX). This strategy can also be applied for a deletion of a gene using the MX cassette (Goldstein and McCusker, 1999) or any developed genetic allele. The inserted [u1] sequences are included within two overlapping oligonucleotides that allow for the inclusion of the desired target de novo within the assembled plasmid. The example shown includes use of the S. pombe HIS5 MX cassette; selection for recircularization of the plasmid is also sufficient. 2. A similar plasmid is also constructed in parallel using in vivo ligation which inserts the [u1] site after the last codon of the gene (as shown) and upstream of the native terminator sequence. For selection purposes, the ADH1-MX cassette was also included. 3. Each created plasmid serves as the template for two PCRs which contain both identical [u1] sites at the proper position; the overlapping homology created between the fragments spans the entire open reading frame of the sample gene (as shown). 4. The newly designed cassette can be amplified by PCR and contains the gene of interest flanked by the two [u1] sites as well as 5’ and 3’ UTR sequences (also referred to as prGeneX and GeneX(t), respectively) for integration (see Figure 4). - Following successful construction of the two separate [u1] tagged sequences, a second round of assembly should be performed using amplified products from each of the original vectors. In this case, while the [u1] could technically provide 23 (or 24) bp of overlapping homology, the entirety of the full gene provides > 1,200 bp of homology to greatly bias the cross-over to occur within this sequence and shift the propensity for assembly to include both [u1] sites as well as the central gene. Both the CDC11 (WT gene) and SHS1 (deletion cassette) were constructed with flanking [u1] sites.
Note: This process could also be achieved using a deletion cassette rather than the WT coding sequence of a gene. Should the tagged cassette include a selectable marker, then construction may be done in a single step since active selection would ensure the presence of both [u1] sites as well as the deletion marker. - For the construction of the Cas9-expression cassette (Figure 3), we demonstrate the use of an additional intermediate plasmid that was built due to the complexity of the final intended product (nine separate DNA sequences to be assembled). Following the individual plasmid assemblies to insert the [u1] (shown) or [u2] target sites, the first intermediate created a vector with a unique restriction site (NotI) between the inducible promoter and generic terminator sequences. Another round of in vivo assembly allowed for the inclusion of the Cas9 gene (in two amplified fragments; amplified from Addgene plasmid #43804) and a C-terminal NLS sequence.
Note: This intermediate integrating vector (Figure 3, #3) would allow for the rapid inclusion of any Cas9 ortholog into the [u1]- or [u2]-containing HIS3 locus in yeast.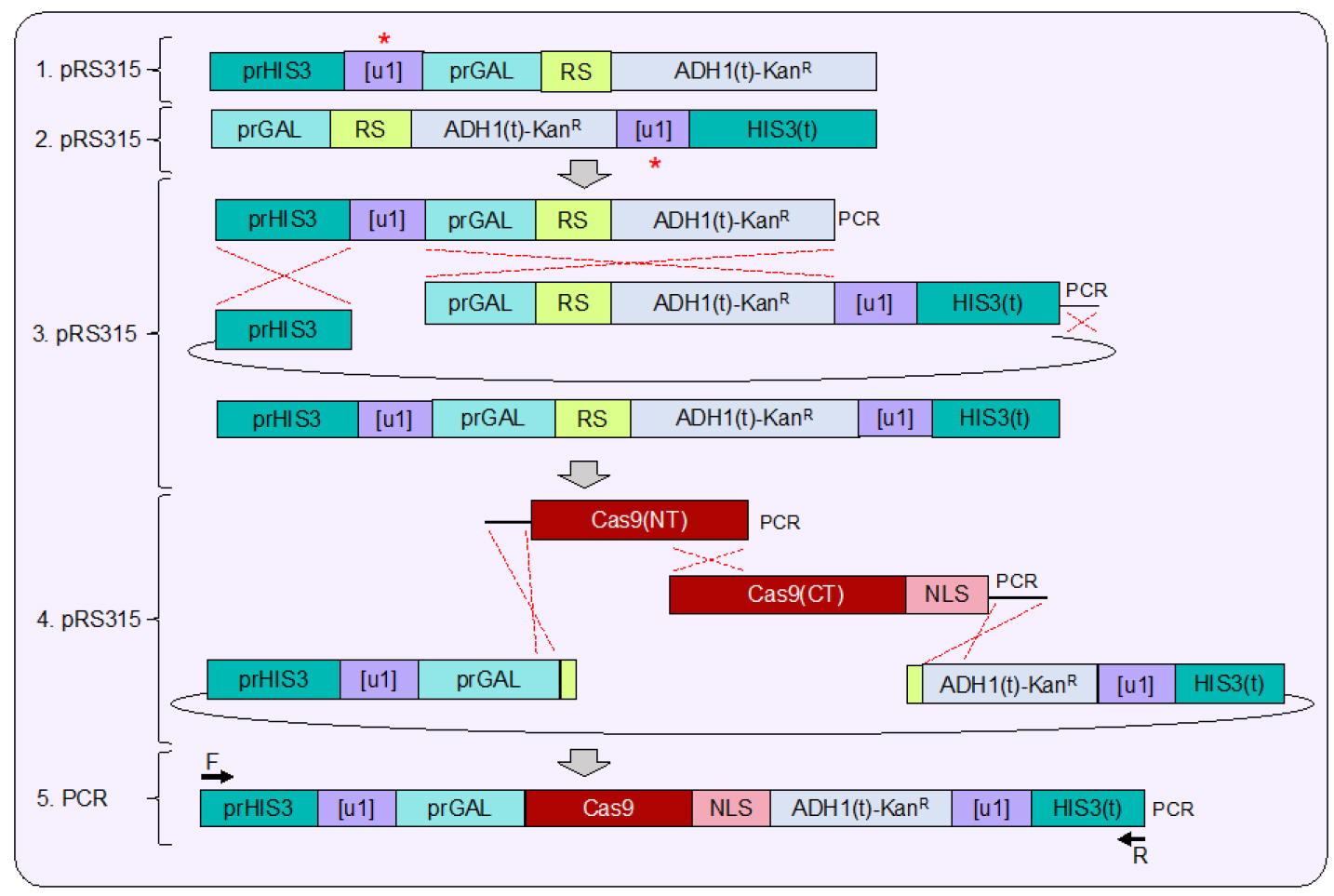
Figure 3. Plasmid assembly of the Cas9 expression cassette. 1. Similar to assembly of the flanking [u1] sites surrounding the sample gene (Figure 2), construction of the [u1] or [u2] flanked expression cassette is done in a stepwise fashion (red asterisks). S. pyogenes Cas9 is under control of the GAL1/10 promoter and is placed downstream of the unique site and the 5’ UTR of the yeast HIS3 gene. A unique restriction site (RS, NotI) was included after the start codon and before the ADH1 terminator sequence. 2. The [u1] or [u2] site was placed after the entire MX terminator sequence but upstream of the yeast HIS3 3’ UTR. 3. Following construction of each separate vector, both fragments were PCR amplified and combined into a larger vector using in vivo ligation to create an intermediate expression vector. 4. Digestion of the unique NotI restriction site allows for a final step of plasmid assembly to insert Cas9 and a NLS (nuclear localization signal), both amplified in two overlapping PCR fragments in-frame with the start codon. 5. Following verification of the entire Cas9 expressing cassette, PCR amplification allows for integration into the yeast genome at the HIS3 locus (see Figure 4).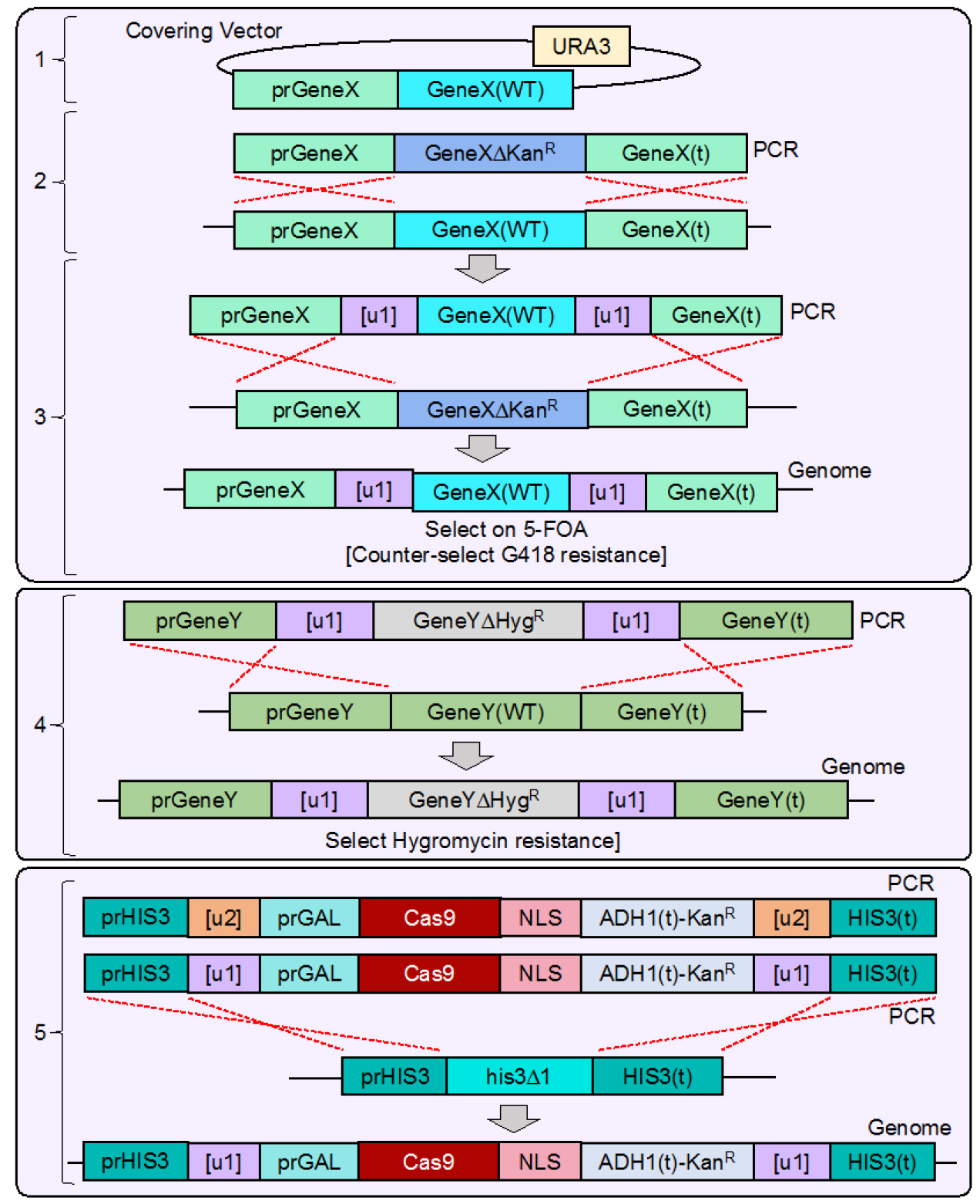
Figure 4. Integration of [u1] flanked gene(s) and Cas9 expression cassette into the yeast genome. As a proof of concept, we chose to pilot this methodology in yeast using two native yeast genes (CDC11 and SHS1) as well as integrate Cas9 at a safe-harbor (HIS3) locus. CDC11 is an essential gene, and our strategy illustrates the additional steps taken to allow for modification and Cas9-based editing of both essential and non-essential genes. 1. WT BY4741 yeast were transformed with a URA3-based covering vector expressing a WT copy of GeneX (CDC11) lacking 3’ UTR sequence. 2. The endogenous GeneX copy was deleted using the standard MX-based deletion cassette and selected on G418-containing medium. 3. The entire [u1]-flanked GeneX construct (Figure 2) was amplified and transformed into the GeneX∆ strain and selected on media containing 5-FOA to counter-select for the URA3-based covering vector. The only viable yeast remaining would have integrated the [u1]-flanked GeneX (WT) copy in place of the KanR cassette resulting in a marker-less modification. Yeast integration events were all confirmed by diagnostic PCR and DNA sequencing following each round of successive modifications. 4. At a separate GeneY locus (SHS1), a deletion cassette was constructed and flanked by [u1] Cas9 sites (similar strategy as GeneX in Figure 2) and transformed into yeast to delete the endogenous GeneY copy. 5. Finally, the entire Cas9-expression cassette (Figure 3) was amplified, transformed into yeast, and integrated at the HIS3 locus (his3∆1), providing the strain G418 resistance. As shown, the final strain contains six identical [u1] target sites. However, an alternative construct was also tested using a [u2] sequence distinct from the [u1] site that flanked the Cas9 cassette allowing for separate and sequential editing at the HIS3 locus. - Following construction and verification of the desired plasmids (Figures 2, 3 bottom), a final PCR amplification allows for the entire designed cassettes to be integrated into the yeast genome (see Procedure C).
- Donor PCRs may be amplified from plasmid or chromosomal preparations with at least 30 bp of flanking homology on either side of the DSB break(s). The PCR fragments (CDC11, SHS1, and HIS3) used as donor DNA for our proof of concept were PCR amplified from an isolated chromosomal DNA prep of WT yeast with several hundred bases of flanking UTR sequence. For amplification of WT HIS3, SF838-1Dα genomic DNA was used.
- The sgRNA-expression plasmid may be constructed using a variety of protocols. The construction of our sgRNA included the following:
- The sgRNA cassette sequence was modeled after (DiCarlo et al., 2013) with the SNR52 promoter, the SUP4 terminator, and included both the [u1] 20 bp target sequence and the required structural RNA sequence (79 bp) for S. pyogenes Cas9. This entire cassette was synthesized de novo from Genscript (Piscataway, NJ).
Note: Numerous methodologies have been developed that allow for construction of the sgRNA plasmid including restriction digest and ligation, in vivo assembly, or Gibson cloning (Laughery et al., 2015; Ryan et al., 2016). - Flanking restriction sites should be included in the synthesized sgRNA cassette (Figure 1C of Finnigan and Thorner, 2016). Our synthesized gene (Genscript) was cloned into pUC57 and contained flanking BamHI and XhoI sites. The high-copy pRS425 yeast plasmid and the [u1] sgRNA plasmid should be digested with both BamHI and XhoI overnight at 37 °C (incubator), linearized fragments separated on a 1% agarose gel, and extracted using a Qiaquick gel extraction kit (QIAGEN).
- Purified DNA should be ligated together at 16 °C for 16 h using T4 DNA ligase (NEB); the reaction halted by incubating at 65 °C for 10 min (performed in PCR block).
- 100 μl of TOP10 competent E. coli should be transformed with 10 μl of the ligation reaction, incubated on ice for 15 min, heat-shocked at 42 °C for 45 sec (water bath), recovered on ice for 2 min, gently mixed with 500 μl of SOC medium (see Recipes), and incubated at 37 °C for 1 h (incubator with slow rotation).
- Cells should be plated onto LB + AMP plates (see Recipes) and incubated overnight at 37 °C. Clonal isolates are to be cultured in liquid LB + AMP medium (overnight in a 37 °C water bath for 14-16 h) and plasmids are extracted using a bacterial plasmid extraction kit (GeneJet). Diagnostic digests followed by DNA sequencing of sgRNA plasmids can be used to confirm proper subcloning from pUC57 to pRS425.
- In order to create the [u2] targeting sgRNA plasmid (which is identical except for the 20 bp target sequence), successive rounds of a modified quick-change PCR (Zheng et al., 2004) can be used to mutate the [u1] sequence into [u2]. (This could be achieved on either the smaller pUC57 vector followed by the aforementioned subcloning steps, or the final pRS425 vector). Briefly, 60 bp oligonucleotides (a forward and reverse primer) should be designed that are complementary and contain a central 1-4 base substitution (the changes do not have to be successive but should be within a relatively close region).
- The entire vector should be PCR amplified using the high-fidelity KOD hot-start polymerase (EMD Millipore) with the recommended conditions, an annealing temperature of 50 °C, and an extension time of 40 sec/Kb. Briefly, a 50 μl reaction (total volume) containing the KOD components (Mg2+, 1x buffer solution, and 1x dNTPs, all provided), template DNA (100-500 ng), oligonucleotides (forward and reverse, final concentration 1 μM each primer), and 1 μl of KOD enzyme should be prepared. An initial hot-start cycle (95 °C, 2 min) followed by cycles consisting of (i) 95 °C, 15 sec, (ii) 50 °C, 30 sec, and (iii) 72 °C, X sec (where X is the total size of the template plasmid divided by 40 sec/Kb). Following 16 total cycles, a final 10 min extension time at 72 °C should be included.
- Following confirmation on a DNA agarose gel, 7.5 μl of the PCR reaction should be digested with 1 μl of DpnI overnight at 37 °C (incubator) with a final reaction volume of 50 μl. Digestion with DpnI is used to destroy the circular (methylated) template yeast plasmid DNA within the PCR reaction in order to remove false positives clones that may propagate in yeast due to template DNA rather than the intended plasmid assembly.
- 2 μl of the DpnI-treated digestion reaction should be transformed into E. coli as previously described.
- Clonal isolates should be confirmed via DNA sequencing.
- The sgRNA cassette sequence was modeled after (DiCarlo et al., 2013) with the SNR52 promoter, the SUP4 terminator, and included both the [u1] 20 bp target sequence and the required structural RNA sequence (79 bp) for S. pyogenes Cas9. This entire cassette was synthesized de novo from Genscript (Piscataway, NJ).
- Following selection of the unique [u1, u2, u3…] artificial sites (Procedure A), as well as the intended strategy to flanking the loci of interest with said sites (Figure 1, bottom), in vivo plasmid assembly (Finnigan and Thorner, 2015) can be used to install the sites at their precise position(s).
- Integration of constructs into the yeast genome
- Our strategy for placement of the [u1]/[u2] flanked DNA sequences into the yeast genome illustrated the ability of both essential (CDC11) and non-essential (SHS1) genes to be manipulated with this methodology (Figure 4). Variations of our step-wise integration protocol could be achieved using different gene deletions or other methods including (i) integration of separate tagged genes into opposite mating types in parallel and subsequent mating, diploid formation, and sporulation (given the manipulated genes include a selectable marker of some sort); (ii) integration vectors such as the pRS300-series (Sikorski and Hieter, 1989), and/or (iii) CRISPR/Cas9 itself to create double stranded breaks and allow for insertion of the [u1]-flanked fragments as donor DNA. Our procedure illustrates one possible strategy to construct the intended yeast strain.
- WT BY4741 yeast should be transformed with a protective ‘covering vector’ (URA3-marked) using a modified lithium acetate protocol (Finnigan and Thorner, 2015) and selected on SD-URA plates.
- A knock-out deletion cassette (Finnigan et al., 2015) for the essential gene (e.g., CDC11) should be PCR amplified (with sufficient 5’ and 3’ UTR flanking sequences–several hundred bps), digested overnight at 37 °C with DpnI, transformed into yeast, and selected on medium containing G418. Additionally, colonies should also be sensitive on plates containing 5-FOA (to counter-select for the URA3-marked covering plasmid) since loss of an essential gene (e.g., cdc11∆) render yeast inviable.
Note: The URA3-marked covering vector must not include any native 3’ UTR, otherwise the deletion cassette may delete the plasmid-borne copy rather than the endogenous gene. - For clarity/simplicity, we will refer to our proof of concept with regards to the CDC11 essential gene, the SHS1 non-essential gene, and Cas9 itself. The [u1]-CDC11-[u1] (essential gene) PCR fragment with flanking UTR (> 300 bp) (Figure 2) should be transformed into cdc11∆ yeast following treatment with DpnI. Since no selectable marker is present on the integrating fragment, yeast should be plated onto medium containing 5-FOA (to select for the restoration of the CDC11 locus and cell viability). All generated yeast strains should be verified by diagnostic PCR and DNA sequencing.
- Second, the SHS1 locus should be deleted using the [u1]-shs1∆::HygR-[u1] with flanking UTR sequence (Figure 2) in the standard fashion, by transforming yeast (from step C4) with the amplified PCR product following DpnI digestion, and selection on medium containing hygromycin.
- Third, the HIS3 locus should be modified with the addition of one (or both) versions of the Cas9-expression cassette–flanked by either the [u1] or [u2] sites. Due to the large size of the entire Cas9 cassette (Figure 3) including the HIS3 5’ and 3’ UTR sequences (500 bp), two amplified PCR products may be generated using primers internal to the Cas9 gene itself (generating at least 100 bp of internal overlap for recombination), treated with DpnI, co-transformed into yeast (from step C5), and selected on medium containing G418. Colonies should also be tested for hygromycin resistance; due to the common promoter and terminator sequences between all the MX-based drug cassettes (Goldstein and McCusker, 1999), it is advisable to confirm all marked loci after each round of selection (by growth phenotypes and possibly by diagnostic PCRs) since there is a propensity for marker ‘swapping’ that may compete with integration at the desired site. The integration vector for the Cas9-expression cassette can target any S. cerevisiae strain harboring some 5’ and 3’ UTR sequences (within 1,000 bp on either side of the HIS3 gene). This integration will also be effective in strains containing an intact HIS3 gene, his3∆1, or his3∆0 yeast.
Note: Confirmation of the final [u1]- and [u2]-flanked strains should include PCR amplification of the three manipulated loci (CDC11, SHS1, and HIS3) with a high-fidelity polymerase, PCR purification, and DNA sequencing.
- Our strategy for placement of the [u1]/[u2] flanked DNA sequences into the yeast genome illustrated the ability of both essential (CDC11) and non-essential (SHS1) genes to be manipulated with this methodology (Figure 4). Variations of our step-wise integration protocol could be achieved using different gene deletions or other methods including (i) integration of separate tagged genes into opposite mating types in parallel and subsequent mating, diploid formation, and sporulation (given the manipulated genes include a selectable marker of some sort); (ii) integration vectors such as the pRS300-series (Sikorski and Hieter, 1989), and/or (iii) CRISPR/Cas9 itself to create double stranded breaks and allow for insertion of the [u1]-flanked fragments as donor DNA. Our procedure illustrates one possible strategy to construct the intended yeast strain.
- CRISPR/Cas9 multiplexed editing in budding yeast using artificial target sites
- A modified yeast transformation protocol is used for Cas9 editing. First, overnight cultures (synthetic drop out media minus uracil, 2% raffinose, 0.2% sucrose, see Recipes) are prepared for the yeast strain containing all six [u1] sites as well as the URA3-marked covering vector expressing WT CDC11 and incubated at 30 °C.
- Yeast should be back-diluted to an OD600 of approximately 0.30 into YPGal (2% galactose) and incubated for 4.5 additional hours at 30 °C to induce Cas9 expression.
- Approximately 10 OD600 of cells should be harvested at 7,300 x g, washed with 0.5 ml water, centrifuged once more at the same speed, and the water should be removed with a sterile pipet.
- Yeast should be resuspended in 0.5 ml of 100 mM sterile lithium acetate, centrifuged at 7,300 x g, and the supernatant should be removed (leaving the cells in the tube).
- A ‘PEG master mix’ should be created (fresh) containing the following: 240 μl of 50% polyethylene glycol (PEG), 36 μl of 1 M lithium acetate, 50 μl of SS DNA (boiled for 10 min and cooled on ice) and a variable amount of sterile water (donor DNA, plasmid DNA, and water should total 34 μl). The mixture (including water) should be vortexed separately and added to the yeast pellet.
- DNA for the Cas9 editing should include the following:
- The sgRNA plasmid (between 1,000-1,500 ng total DNA).
- Donor DNA (for each edited locus) as amplified PCR fragments (1,000-1,500 ng total DNA, varied based on length of PCR product).
- The sgRNA plasmid (between 1,000-1,500 ng total DNA).
- Control reactions for Cas9 editing should include (i) empty pRS425 vector with no sgRNA–this serves as an upper bound for the total number of transformants even in the absence of donor DNA of any kind; (ii) cultured yeast in YPDex (2% dextrose) (see Recipes) rather than galactose–however, this may yield a significantly different number of transformed colonies given the difference in metabolic activity and total cell number, and/or (iii) inclusion of the sgRNA plasmid and no donor DNA of any kind (sterile water)–this causes Cas9-induced double stranded breaks but requires any surviving yeast to perform NHEJ at each DSB location (two controls are demonstrated in Figure 5).
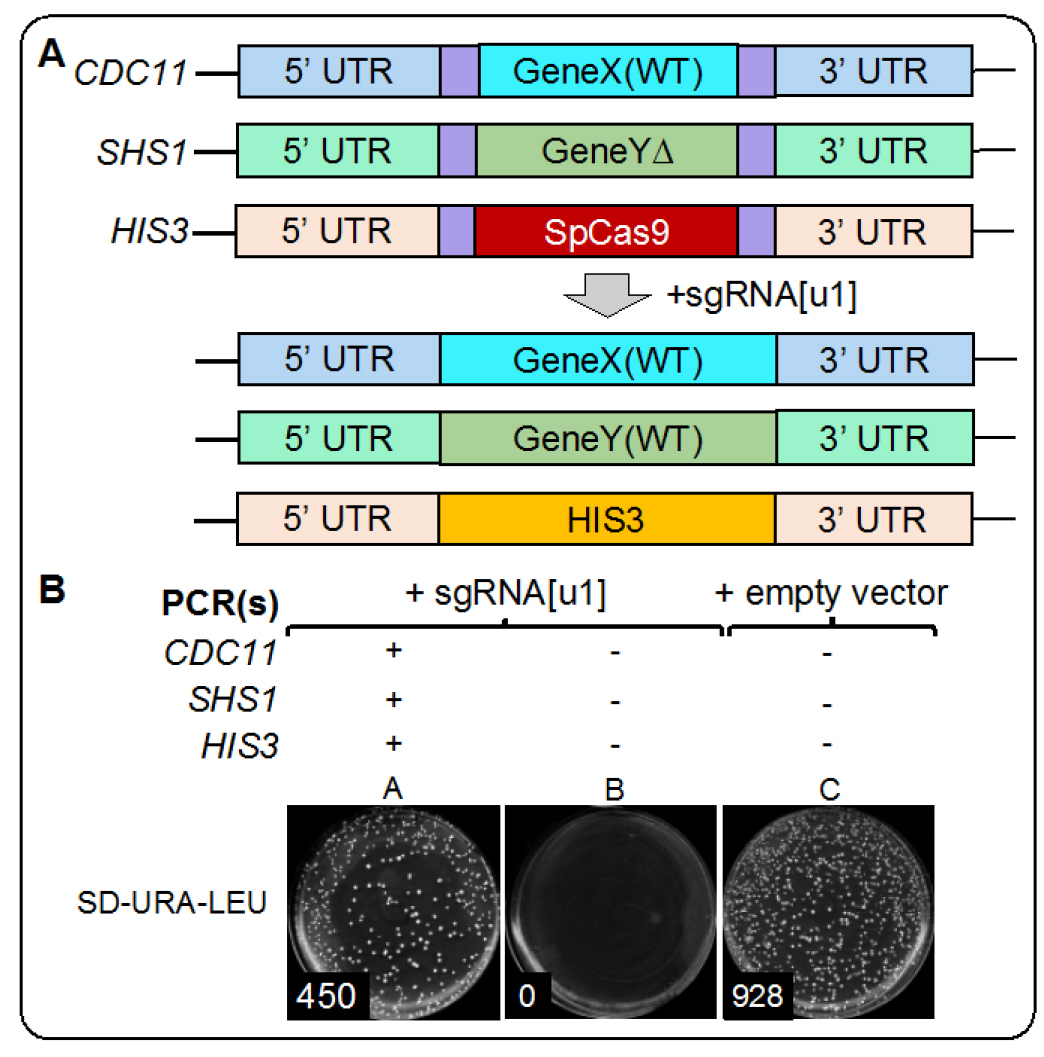
Figure 5. Example results from Cas9-based editing in vivo. A. Schematic of the generated yeast strain with six identical [u1] artificial sites flanking three loci (GeneX, GeneY, and Cas9 at the HIS3 locus) and, after addition of the sgRNA for [u1] and donor DNA for each locus, the expected replacement alleles (WT) for all edited genomic locations. B. As a proof of concept, we provide example data from our Cas9 mCAL editing in vivo. Plate A includes yeast transformed with the sgRNA and all three donor PCR fragments. Plate B includes sgRNA but no donor DNA. Plate C includes an empty plasmid (identical to the guide RNA plasmid backbone) and no donor DNA. Because DSBs are lethal in yeast, Plate B results in no viable colonies whereas inclusion of the donor DNA allows for repair of each manipulated locus and a significant number of colonies. The provided example does not use any selection for integrated donor DNA and merely selects for the presence of a covering vector (URA3) and the single sgRNA plasmid (LEU2). - Following addition of the PEG master mixture to yeast, the appropriate amount of DNA (plasmid and PCRs) should be added, and the tube vortexed at maximum setting for 10 sec to achieve full resuspension.
- Yeast should be heat shocked at 42 °C for 45 min, centrifuged at 7,300 x g for 1 min, and the PEG mixture should be removed with a pipet.
- Yeast should be resuspended in fresh YPGal liquid and placed at 30 °C overnight (14-16 h) to recover.
- Following recovery, cells (and the YPGal liquid) should be plated directly onto SD-URA-LEU plates and incubated at 30 °C for three days prior to imaging. Three example conditions from our proof of concept are demonstrated in Figure 5. Plate A. Yeast were transformed with the sgRNA [u1] plasmid and donor DNA for all three loci. Plate B. Yeast were transformed with the sgRNA [u1] plasmid and no donor DNA. Plate C. Yeast were transformed with an empty pRS425 vector and no donor DNA. The total number of colonies is recorded on each plate (Figure 5 bottom). Cas9-induced DSBs render cells inviable (Figure 5B), but the inclusion of donor DNA for all three loci allows for subsequent repair by homologous recombination, and rescue of cell viability (Figure 5A).
Note: The proof of concept illustrates use of Cas9 editing with the artificial sites with no selection for markers (lack of uracil selects for the covering vector whereas lack of leucine selects for the presence of the [u1] sgRNA). However, selection/markers can be used if needed.
- A modified yeast transformation protocol is used for Cas9 editing. First, overnight cultures (synthetic drop out media minus uracil, 2% raffinose, 0.2% sucrose, see Recipes) are prepared for the yeast strain containing all six [u1] sites as well as the URA3-marked covering vector expressing WT CDC11 and incubated at 30 °C.
- Verification of editing genome sequences
- Confirmation of Cas9 editing of yeast should be performed by first preparing genomic DNA from clonal isolates (Amberg et al., 2006).
- Diagnostic PCRs should be performed using genomic DNA as a template to test for both the identity and sizes of the manipulated loci (in our case, CDC11, SHS1, and HIS3) to assess whether donor DNAs were properly integrated into the genome. As an example, removal of the flanking [u1] sites for CDC11 (and replacement with the native gene) displayed a small shift upon loss of the artificial site when separated on a 2% agarose DNA gel. Similarly, the integration of the WT SHS1 gene (in place of the deletion) allowed for PCR combinations that provided evidence of proper integration.
- Finally, manipulated loci should be amplified once more, purified, and sent for DNA sequencing.
Note: As shown, the example strategy illustrates the Cas9-expression cassettes flanked by [u1] sites. We also tested the sequential elimination of Cas9 after editing of additional loci by construction of a [u2]-flanked Cas9 cassette along with the appropriate guide RNA and donor DNA (Finnigan and Thorner, 2016). In this scenario, consecutive transformations would allow for removal of the Cas9 gene at a later time.
- Confirmation of Cas9 editing of yeast should be performed by first preparing genomic DNA from clonal isolates (Amberg et al., 2006).
Data analysis
For all stages of plasmid and yeast strain construction (Procedures A-E), diagnostic PCRs and final confirmation via DNA sequencing were performed to assess that all assembled DNA fragments (plasmid or genome) were generated as expected. An online DNA alignment program (Biology Workbench, San Diego Supercomputer Center) was used to analyze Sanger sequences. For Figure 5, colonies were counted using a sectoring method; independent experiments were performed in triplicate–only a representative plate for each trial is presented (http://www.g3journal.org/content/6/7/2147.long) (Finnigan and Thorner, 2016).
Notes
Our system includes a number of significant improvements and novel uses for CRISPR/Cas9-based gene editing in budding yeast.
- The use of a single sgRNA to target multiple genomic loci (reduces the need for multiple sgRNA constructs).
- Construction of an initial yeast strain pre-programmed with all unique sites flanking all genes of a complex or pathway would allow for massively parallel future multiplexing and strain generation via Cas9; all combinations would be possible because WT copies of each unmodified gene could easily be repaired (as we have shown).
- Allows for seamless excision of the Cas9 gene itself either in conjunction with modification of other loci or sequentially at a future time.
- Programming of the artificial sites on either side (flanking) the locus of interest ensures excision of the entire gene/marker/cassette and helps prevent promiscuous cross-over should a single allele or similar mutant need to be integrated in place of the WT copy.
- This process can be used to edit essential genes.
- This method could be applied to deletion or modification of entire chromosome segments, groups of genes, or multiple genes.
- Cas9-based editing allows for marker-less integration (the plasmid harboring the guide RNA can be easily lost with no selective pressure).
- Reduction/elimination off-target effects–since there is absolute control over the sequence that will be targeted within the genome, this should aid in optimizing editing while reducing recruitment of Cas9 to similar or other genomic loci.
- As a consequence of having the exact same unique artificial site multiple times within a genome, it would allow for a comprehensive screen/search for local epigenetic or positional effects that might alter Cas9 editing since the sgRNA and the target sequence would be identical across the genome.
- Aid in synthetic genome engineering–inclusion of Cas9 target site(s) would allow future editing, chromosome splitting, rearrangements, etc., across the entire genome.
Recipes
- YPD liquid media
1% yeast extract
2% peptone
2% dextrose
Autoclaved - YPGal
1% yeast extract
2% peptone
2% galactose
YP autoclaved, galactose filter sterilized, mixed - SOC medium
2% tryptone
0.5% yeast extract
10 mM NaCl
2.5 mM KCl
1 mM MgCl2
1 mM MgSO4
20 mM glucose final concentration - YPD plates (with appropriate drug included if needed)
1% yeast extract
2% peptone
2% dextrose
20 g/L Bacto agar
Autoclaved
Liquid drug stocks added once media cooled < 55 °C - Synthetic drop-out media (e.g., -URA-LEU)
1.7 g yeast nitrogen base minus ammonium sulfate and minus amino acids
5.0 g ammonium sulfate
1.1 g of ‘almost complete’ amino acid mixture
Remaining amino acid mixture (All from Sigma-Aldrich, methionine 150 mg/L; lysine 180 mg/L; histidine 60 mg/L
Note: If needed, leucine 260 mg/L and uracil 20 mg/L.
10 mg/L tryptophan (filter sterilized, not autoclaved)
2% dextrose
20 g/L Bacto agar
Sterile water and agar are mixed and autoclaved separately from remaining component - 5-FOA plates
Synthetic media components (appropriate amino acid drop-out, if any)
0.5 g/L 5-FOA
0.5 g/L uracil
Heat all components (separate from water/agar mixture) to 75 °C for 30 min, filter sterilize and combine with autoclaved water/agar mixture - LB plates (with appropriate drug included)
1% tryptone
0.5% yeast extract
15 g/L agar
10 g/L NaCl
4 mM NaOH - TAE buffer (1x final concentration), pH 8.4
40 mM Tris (1 M stock solution)
20 mM glacial acetic acid
1 mM EDTA (0.5 M stock solution, pH 8.0)
Acknowledgments
This project was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20 GM103418 to G.C.F. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. This work was also supported by an Innovative Research Award (to G.C.F.) from the Johnson Cancer Research Center at Kansas State University. This work was supported by an Undergraduate Research Award (to R.M.G.) from the College of Arts and Sciences at Kansas State University. This protocol was modified and adapted from (Finnigan and Thorner, 2016). We would like to thank Jeremy Thorner (Univ. of California, Berkeley) for useful advice and comments.
References
- Amberg, D. C., Burke, D. J. and Strathern, J. N. (2006). Yeast DNA isolation: midiprep. CSH Protoc 2006(1).
- Brachmann, C. B., Davies, A., Cost, G. J., Caputo, E., Li, J., Hieter, P. and Boeke, J. D. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14(2): 115-132.
- Cho, S. W., Kim, S., Kim, Y., Kweon, J., Kim, H. S., Bae, S. and Kim, J. S. (2014). Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24(1): 132-141.
- Demirci, Y., Zhang, B. and Unver, T. (2017). CRISPR/Cas9: An RNA-guided highly precise synthetic tool for plant genome editing. J Cell Physiol.
- DiCarlo, J. E., Chavez, A., Dietz, S. L., Esvelt, K. M. and Church, G. M. (2015). Safeguarding CRISPR-Cas9 gene drives in yeast. Nat Biotechnol 33(12): 1250-1255.
- DiCarlo, J. E., Norville, J. E., Mali, P., Rios, X., Aach, J. and Church, G. M. (2013). Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41(7): 4336-4343.
- Doudna, J. A. and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213): 1258096.
- Estrela, R. and Cate, J. H. (2016). Energy biotechnology in the CRISPR-Cas9 era. Curr Opin Biotechnol 38: 79-84.
- Finnigan, G. C., Takagi, J., Cho, C. and Thorner, J. (2015). Comprehensive genetic analysis of paralogous terminal septin subunits Shs1 and Cdc11 in Saccharomyces cerevisiae. Genetics 200(3): 821-841.
- Finnigan, G. C. and Thorner, J. (2015). Complex in vivo ligation using homologous recombination and high-efficiency plasmid rescue from Saccharomyces cerevisiae. Bio Protoc 5(13).
- Finnigan, G. C. and Thorner, J. (2016). mCAL: a new approach for versatile multiplex action of Cas9 using one sgRNA and loci flanked by a programmed target sequence. G3 (Bethesda) 6(7): 2147-2156.
- Goldstein, A. L. and McCusker, J. H. (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15(14): 1541-1553.
- Jensen, E. D., Ferreira, R., Jakociunas, T., Arsovska, D., Zhang, J., Ding, L., Smith, J. D., David, F., Nielsen, J., Jensen, M. K. and Keasling, J. D. (2017). Transcriptional reprogramming in yeast using dCas9 and combinatorial gRNA strategies. Microb Cell Fact 16(1): 46.
- Jiang, W., Bikard, D., Cox, D., Zhang, F. and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31(3): 233-239.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Laughery, M. F., Hunter, T., Brown, A., Hoopes, J., Ostbye, T., Shumaker, T. and Wyrick, J. J. (2015). New vectors for simple and streamlined CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Yeast 32(12): 711-720.
- Men, K., Duan, X., He, Z., Yang, Y., Yao, S. and Wei, Y. (2017). CRISPR/Cas9-mediated correction of human genetic disease. Sci China Life Sci 60(5): 447-457.
- Rothman, J. H. and Stevens, T. H. (1986). Protein sorting in yeast: mutants defective in vacuole biogenesis mislocalize vacuolar proteins into the late secretory pathway. Cell 47(6): 1041-1051.
- Ryan, O. W. and Cate, J. H. (2014). Multiplex engineering of industrial yeast genomes using CRISPRm. Methods Enzymol 546: 473-489.
- Ryan, O. W., Poddar, S. and Cate, J. H. (2016). CRISPR-Cas9 genome engineering in Saccharomyces cerevisiae cells. Cold Spring Harb Protoc 2016(6): pdb prot086827.
- Sasano, Y., Nagasawa, K., Kaboli, S., Sugiyama, M. and Harashima, S. (2016). CRISPR-PCS: a powerful new approach to inducing multiple chromosome splitting in Saccharomyces cerevisiae. Sci Rep 6: 30278.
- Sikorski, R. S. and Hieter, P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122(1): 19-27.
- Si, T., Chao, R., Min, Y., Wu, Y., Ren, W. and Zhao, H. (2017). Automated multiplex genome-scale engineering in yeast. Nat Commun 8: 15187.
- Sorek, R., Lawrence, C. M. and Wiedenheft, B. (2013). CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem 82: 237-266.
- Zhang, X. H., Tee, L. Y., Wang, X. G., Huang, Q. S. and Yang, S. H. (2015). Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4: e264.
- Zheng, L., Baumann, U. and Reymond, J. L. (2004). An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32(14): e115.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Giersch, R. M. and Finnigan, G. C. (2017). Method for Multiplexing CRISPR/Cas9 in Saccharomyces cerevisiae Using Artificial Target DNA Sequences. Bio-protocol 7(18): e2557. DOI: 10.21769/BioProtoc.2557.
Category
Microbiology > Microbial genetics > DNA > Chromosomal
Molecular Biology > DNA > DNA cloning
Molecular Biology > DNA > Chromosome engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.