- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9 Gene Editing in the Marine Diatom Phaeodactylum tricornutum

Published: Vol 7, Iss 15, Aug 5, 2017 DOI: 10.21769/BioProtoc.2442 Views: 14629
Reviewed by: Dennis NürnbergVera Karolina SchoftAgnieszka Zienkiewicz
Original research article
The authors used this protocol in:

May 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols



Abstract
The establishment of the CRISPR/Cas9 technology in diatoms (Hopes et al., 2016; Nymark et al., 2016) enables a simple, inexpensive and effective way of introducing targeted alterations in the genomic DNA of this highly important group of eukaryotic phytoplankton. Diatoms are of interest as model microorganisms in a variety of areas ranging from oceanography to materials science, in nano- and environmental biotechnology, and are presently being investigated as a source of renewable carbon-neutral fuel and chemicals. Here we present a detailed protocol of how to perform CRISPR/Cas9 gene editing of the marine diatom Phaeodactylum tricornutum, including: 1) insertion of guide RNA target site in the diatom optimized CRISPR/Cas9 vector (pKS diaCas9-sgRNA), 2) biolistic transformation for introduction of the pKS diaCas9-sgRNA plasmid to P. tricornutum cells and 3) a high resolution melting based PCR assay to screen for CRISPR/Cas9 induced mutations.
Keywords: CRISPR/Cas9 technologyBackground
The CRISPR/Cas9 system has proven to be a very efficient and successful genome editing system in a number of eukaryotic organisms, now also including microalgae (Hopes et al., 2016; Nymark et al., 2016; Shin et al., 2016). The CRISPR/Cas9 system includes a guide RNA (gRNA) and a nuclease called Cas9 (Sander and Joung, 2014). These two molecules form a complex where the gRNA directs the complex to the target of interest. The Cas9 nuclease induces double strand brakes at the target site that can be repaired by nonhomologous end joining (NHEJ) which can result in indel mutations, or via the homology-directed repair (HDR) pathway that can be exploited to create defined alterations of the DNA. The presented protocol is the first to describe a step-by-step procedure for applying the CRISPR/Cas9 system to create gene-targeted mutations (indels ranging from one to hundreds of nucleotides) in one of the main diatom model species, P. tricornutum.
Materials and Reagents
- Pipette tips, 1,000 µl (SARSTEDT, catalog number: 70.762.100 )
- Pipette tips, 200 µl (SARSTEDT, catalog number: 70.760.502 )
- Pipette tips, 20 µl (Biosphere® Tip 20 µl neutral) (SARSTEDT, catalog number: 70.1116.200 )
- Filter tips, 20 µl (Biosphere® Fil. Tip 20 µl neutral) (SARSTEDT, catalog number: 70.1116.210 )
- 24 or 48-cell multiwell cell culture plates, flat bottom, TC treated (VWR, catalog number: 734-2325 or 734-2326 )
- 1.5 ml tube
- Parafilm
- 50 ml centrifuge tube (SARSTEDT, catalog number: 62.547.254 )
- 0.2 µm sterile filter
- Macrocarriers (Bio-Rad Laboratories, catalog number: 1652335 )
- 1,550 psi rupture discs (Bio-Rad Laboratories, catalog number: 1652331 )
- Stopping screens (Bio-Rad Laboratories, catalog number: 1652336 )
- Phaeodactylum tricornutum cells (NCMA Bigelow Laboratory for Ocean Sciences, Bigelow, catalog number: CCMP2561 starter culture)
- Competent DH5α E.coli cells (‘home-made’ RbCl competent cells [efficiency ≥ 1.0 x 106 cfu/µg])
- pKS diaCas9-sgRNA plasmid (Addgene, catalog number: 74923 )
- pAF6 plasmid (Falciatore et al., 1999) containing the ShBle gene conferring resistance to zeocin
- BsaI-HF restriction endonuclease (New England Biolabs, catalog number: R3535S )
- Complementary oligos (24 nt) with 5’ TCGA and AAAC overhangs (for creation of the adapter for targeting the gene of interest). Custom DNA oligos can be ordered from Sigma-Aldrich
- Wizard® SV Gel and PCR Clean-Up Kit (Promega, catalog number: A9282 )
- T4 DNA ligase buffer (New England Biolabs, catalog number: M0202S )
- ExTaq DNA polymerase and buffer system (AH Diagnostics) (Takara Bio, catalog number: RR001A )
- QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27106 )
- 50% (v/v) seawater plates supplemented with f/2-Si, 1% (w/v) agar plates
- Tungsten M10 or M17 microcarriers (Bio-Rad Laboratories, catalog number: 1652266 or 1652267 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C7902 )
- Spermidine (Sigma-Aldrich, catalog number: S2626 )
- 70% (v/v) and 100% EtOH
- 70% (v/v) isopropanol
- 50% (v/v) seawater plates supplemented with f/2-Si, 1% (w/v) agar plates, 100 µg/ml zeocin
- Zeocin (InvivoGen, catalog number: ant-zn-5p )
- TOPO® TA Cloning® Kit for Sequencing (Thermo Fisher Scientific, InvitrogenTM, catalog number: 450030 )
- LightCycler 480 High Resolution Melting Master Kit (Roche Molecular Systems, catalog number: 04909631001 )
- TritonTM X-100 (Sigma-Aldrich, catalog number: X100 )
- Trizma® base (Sigma-Aldrich, catalog number: T6066 )
- Ethylenediaminetetraacetate acid disodium salt (EDTA) (Sigma-Aldrich, catalog number: E5134 )
- Pancreatic peptone (VWR, catalog number: 26208.297 )
- Bacto yeast extract (BD, BactoTM, catalog number: 212750 )
- Bacteriological agar (VWR, catalog number: 84609.5000 )
- Sodium chloride (NaCl) (Merck, catalog number: 106404 )
- Lysis buffer (Buffer for lysis of P. tricornutum cells) (see Recipes)
- LB medium (see Recipes)
- LB agar plates (see Recipes) containing 100 µg/ml ampicillin
- f/2 growth medium with natural (or artificial) seawater (see Recipes) (Guillard, 1975)
Equipment
- NanoDrop ND-1000 or ND-2000 spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 1000 or NanoDropTM 2000 )
- Heating block (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 88870005 )
- Water bath (Grant Instruments)
- Micropipettes (Eppendorf, model: Eppendorf Research® , variable volume)
- Table top centrifuges (Fisher Scientific, model: accuspinTM Micro 17R , catalog number: 13-100-676)
- Incubator shaker (Eppendorf, New BrunswickTM, model: Innova® 44 , catalog number: M1282-0002)
- Sterile bench (Thermo Fisher Scientific, Thermo ScientificTM, model: Holten Horizontal Laminar Airflow )
- PCR thermal cycler (Bio-Rad Laboratories, model: T100TM, catalog number: 1861096 )
- LightCycler® 96 Real-Time PCR system (Roche Molecular Systems, catalog number: 05815916001 )
- Biolistic PDS-1000/He Particle Delivery System (Bio-Rad Laboratories, catalog numbers: 165-2257 and 165-2250LEASE to 165-2255LEASE )
- Vortex
- Autoclave
- Refrigerator
Software
- LightCycler® 96 Software Version 1.1 (Roche Molecular Systems)
Procedure
- Selection of Cas9-target sites
- Use a custom made or publicly available CRISPR/Cas9 target finding tool (e.g., PhytoCRISP-Ex [Rastogi et al., 2016]) to identify Cas9 target sites (N20-NGG) with low or no homology to other genomic loci. The gene of interest should be sequenced before choosing the target site, or checked for polymorphisms using RNAseq data, because unpublished single nucleotide polymorphism is a frequent finding in the P. tricornutum genome.
- Order 24 nt long oligos with 20 complementary nt and 5’ TCGA and AAAC overhangs for creation of the adapter for targeting the gene of interest (Figure 1).
Optional: Length of the target site can be ± 2 bp.
Figure 1. Example of adapter with 5’ TCGA and AAAC overhangs
- Use a custom made or publicly available CRISPR/Cas9 target finding tool (e.g., PhytoCRISP-Ex [Rastogi et al., 2016]) to identify Cas9 target sites (N20-NGG) with low or no homology to other genomic loci. The gene of interest should be sequenced before choosing the target site, or checked for polymorphisms using RNAseq data, because unpublished single nucleotide polymorphism is a frequent finding in the P. tricornutum genome.
- Ligation of adapter for target of interest into the pKS diaCas9-sgRNA plasmid
- Exploit the two BsaI restriction sites that are placed immediately 5’- to the sgRNA in the vector (Figure 2) to ligate the adapter with 5’-TCGA and 5’-AAAC overhangs into the pKS diaCas9-sgRNA plasmid (Nymark et al., 2016). Digest 2 µg of pKS diaCas9-sgRNA plasmid with 1 µl BsaI-HF (20,000 U/ml, NEB) in a reaction with 5 µl 10x CutSmart buffer (NEB) and DNase-free water to a total reaction volume of 50 µl. Incubate for ≥ 2 h or overnight at 37 °C to ensure complete digestion of the plasmid DNA at both BsaI restriction sites producing incompatible ends. Heat inactivate the BsaI-HF enzyme at 65 °C for 20 min. Clean up the digestion reaction with Wizard® SV Gel and PCR Clean-Up Kit (Promega). Estimate the linearized plasmid concentration by measuring the absorbance at 260 nm using a NanoDrop spectrophotometer.
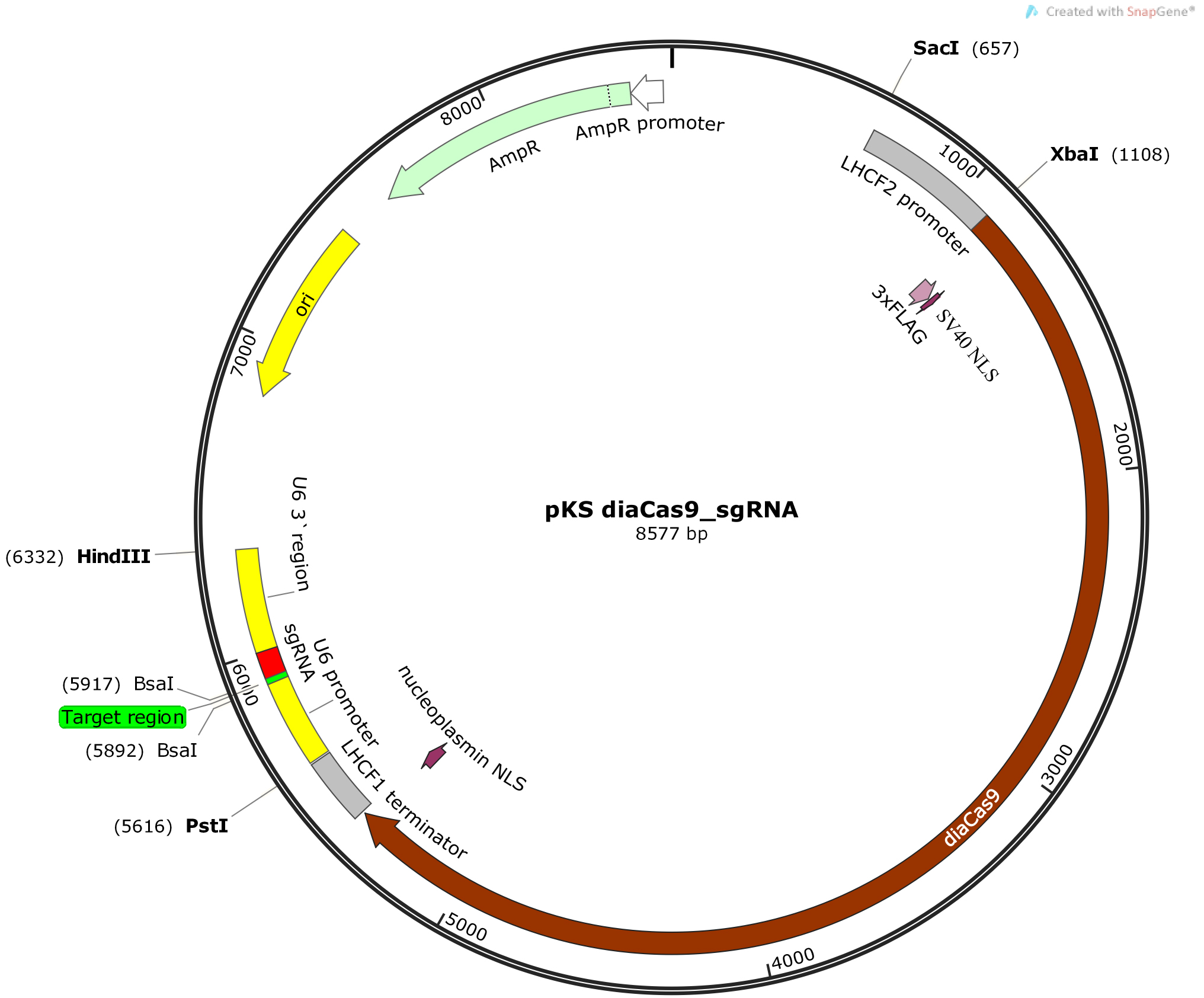
Figure 2. Schematic illustration of the pKS diaCas9-sgRNA plasmid. A diatom codon optimized Cas9 is placed under control of a promoter for a fucoxanthin chlorophyll a/c binding gene (LHCF2), and the expression of the sgRNA is driven by the P. tricornutum U6 promoter. Two BsaI cutting sites are located within the sgRNA cassette to enable insertion of a small adapter for targeting the gene of interest (Nymark et al., 2016). - Anneal the oligos by setting up an annealing reaction containing 1 µg of each oligo, 5 µl 10x T4 ligase buffer (NEB), and DNase-free water to a total reaction volume of 50 µl. Incubate the annealing reaction at 85 °C for 10 min in a heating block for disruption of any secondary structures within each oligo. Slowly cool down the reaction to room temperature (~60-90 min) by turning off the heating block while leaving the tube with the annealing reaction in the heating block. Slow cooling facilitates hybridization as new hydrogen bonds form between the complementary oligos. (Alternatively the annealing reaction can be performed in a thermocycler with a 1 °C/min temperature decrease to room temperature following the 10 min incubation at 85 °C).
- Ligate the adapter into the linearized pKS diaCas9-sgRNA plasmid using a molar vector to insert ratio of 1:20. Set up a ligation reaction using 1 µl T4 DNA ligase (400,000 U/ml; NEB) and 2 µl 10x T4 DNA ligase buffer (NEB) in a total reaction volume of 20 µl for 1 h at 16 °C. Heat inactivate the reaction for 10 min at 65 °C.
- Chill on ice, and transform 5 µl of the ligation reaction into competent cells (e.g., competent DH5α E.coli cells).
- Perform PCR colony screening of the transformed E. coli cells. To identify a pKS diaCas9-sgRNA plasmid containing the insert of interest, use one of the oligos used for creation of the adapter as forward primer in combination with a vector specific primer as reverse primer (e.g., the universal M13 reverse [-29] primer [5’-CAG GAA ACA GCT ATG ACC-3’]) in the PCR reaction.
Note: Screening of 4-8 bacterial colonies is usually sufficient to identify a pKS diaCas9-sgRNA plasmid containing the adapter. - Isolate pKS diaCas9-sgRNA plasmids (e.g., QIAprep Spin Miniprep Kit [QIAGEN]) containing the correct insert from the transformed E.coli cells. Relatively high concentrations (> 400-500 ng/µl) is preferable for coating of the tungsten particles during the preparations for biolistic transformation of the diatom cells.
Note: To achieve such concentrations it might be necessary to pellet 10-15 ml of bacterial culture, since the yield of the pKS diaCas9-sgRNA plasmid is often low.
- Exploit the two BsaI restriction sites that are placed immediately 5’- to the sgRNA in the vector (Figure 2) to ligate the adapter with 5’-TCGA and 5’-AAAC overhangs into the pKS diaCas9-sgRNA plasmid (Nymark et al., 2016). Digest 2 µg of pKS diaCas9-sgRNA plasmid with 1 µl BsaI-HF (20,000 U/ml, NEB) in a reaction with 5 µl 10x CutSmart buffer (NEB) and DNase-free water to a total reaction volume of 50 µl. Incubate for ≥ 2 h or overnight at 37 °C to ensure complete digestion of the plasmid DNA at both BsaI restriction sites producing incompatible ends. Heat inactivate the BsaI-HF enzyme at 65 °C for 20 min. Clean up the digestion reaction with Wizard® SV Gel and PCR Clean-Up Kit (Promega). Estimate the linearized plasmid concentration by measuring the absorbance at 260 nm using a NanoDrop spectrophotometer.
- Biolistic transformation for introduction of the pKS diaCas9-sgRNA plasmid to P. tricornutum cells
Note: P. tricornutum cells can be grown under a wide range of light and temperature conditions. Suggested growth condition: 16 h photoperiod, 22 °C, 65 µmol m-2 sec-1.- DAY 1
- Harvest P. tricornutum cells that are in the exponential growth phase by centrifugation (4,500 x g, 10 min). Remove the supernatant and resuspend the cells in f/2 medium to obtain a concentration of 1 x 108 cells/ml. Spread 500 µl of the high-density cell suspension on 50% (v/v) seawater 1% (w/v) agar plates supplemented with f/2-Si. Allow the plate with the cell suspension to dry in a sterile hood (~15-30 min).
Note: The P. tricornutum cells should be kept in exponential growth phase (cell density ~0.2-2 x 106 cells/ml) for approx. 1-2 weeks before performing the biolistic transformation. Cell numbers can be checked by manual counting using a counting chamber or a flow cytometer. As long as the cells have been well maintained during the period before performing the biolistic transformation, the number of cells on the day of harvesting does not seem to be of importance for the transformation efficiency. For practical reasons cell numbers of approx. 0.5-1.5 x 106 cells/ml are preferable before up-concentration of the cell density as described above. - Transfer the agar plates with P. tricornutum cells to the growth room (16 h photoperiod, 22 °C, 65 µmol m-2 sec-1) for overnight storage.
- Harvest P. tricornutum cells that are in the exponential growth phase by centrifugation (4,500 x g, 10 min). Remove the supernatant and resuspend the cells in f/2 medium to obtain a concentration of 1 x 108 cells/ml. Spread 500 µl of the high-density cell suspension on 50% (v/v) seawater 1% (w/v) agar plates supplemented with f/2-Si. Allow the plate with the cell suspension to dry in a sterile hood (~15-30 min).
- DAY 2
Note: Preparation and coating of microcarriers, and washing/sterilization of all materials and equipment necessary for the biolistic bombardment procedure, were performed exactly as described in the Biolistic PSD-1000/He Particle Delivery System manual (Bio-Rad Laboratories).- Vortex a 50 µl aliquot containing 3 mg washed microcarriers (Tungsten M10 or M17 [Bio-Rad Laboratories]) for 5 min to disrupt agglomerated particles.
- Continue vortexing while adding in the following order 2.5 µg pKS diaCas9_sgRNA and 2.5 µg pAF6 plasmid DNA (maximum total volume of plasmid DNA ≤ 10 µl), 50 µl 2.5 M CaCl2 and 20 µl 0.1 M spermidine (free base, tissue culture grade). Continue vortexing for another 3 min.
- Allow the microcarriers to settle for 1 min.
- Pellet the tungsten particles by briefly (~2 sec) spinning the tube in a microfuge.
- Remove supernatant.
- Add 140 µl of 70% (v/v) EtOH and remove it again by pipetting (no mixing or centrifugation).
- Add 140 µl of 100% EtOH and remove it again by pipetting (no mixing or centrifugation).
- Add 50 µl of 100% EtOH.
- Resuspend by carefully tapping the tube several times.
- While vortexing at a low speed, remove 10 µl of the coated tungsten particles and spread the particles at the center of a macrocarrier using a pipette with a 10-200 µl tip. Repeat this step four times (loading in total five macrocarriers). After the ethanol has evaporated from the macrocarriers assemble all parts necessary for performing the bombardment according to the manufacturer’s instructions (Biolistic PSD-1000/He Particle Delivery System manual, Bio-Rad Laboratories). Use 1,550 psi rupture discs, and sterilize the rupture discs by dipping them briefly (~1-2 sec) in 70% (v/v) isopropanol just prior to insertion in the Retaining Cap.
- Place the target shelf with the agar plate containing P. tricornumtum cells 6 cm below the stopping screen (level 2) in the bombardment chamber.
- Set the vacuum level in the bombardment chamber to 26.5 psi.
- Bombard the same agar plate five times. After each shot, mark the impact area, and change the position of the plate to ensure that the subsequent shots are delivered on another part of the plate.
- Return the plate to the growth room for a minimum time of 24 h.
- Vortex a 50 µl aliquot containing 3 mg washed microcarriers (Tungsten M10 or M17 [Bio-Rad Laboratories]) for 5 min to disrupt agglomerated particles.
- DAY 3
- After a minimum incubation time of 24 h, transfer the algae to selection plates (50% [v/v] seawater plates supplemented with f/2-Si, 1% [w/v] agar plates, 100 µg/ml zeocin). Add 1 ml f/2 to the plate used for microparticle bombardment. Wash off the cells and transfer cells suspension (using a 1 ml pipette) to two selection plates.
- Wash the plate used for microparticle bombardment with an additional 0.5 ml f/2 medium. Transfer the cell suspension to a third selection plate.
Note: Spreading of the cell suspension to three selection plates is performed to lower the cell density on the selection plates and to increase the sensitivity to zeocin, thereby avoiding growth of non-transformed cells. - Dry the plates in the sterile bench (10-15 min)
- Seal the plates with Parafilm and incubate them in the growth room (growth conditions as above) for 2-4 weeks. (Expected number of transformed colonies: ~10-100)
- DAY 1
- Screening for CRISPR/Cas9 induced mutations (see Figure 3 for overview of screening procedure)
Note: To avoid PCR product contamination, filter pipette tips should be used during preparation for and performance of High Resolution Melting (HRM) Analyses.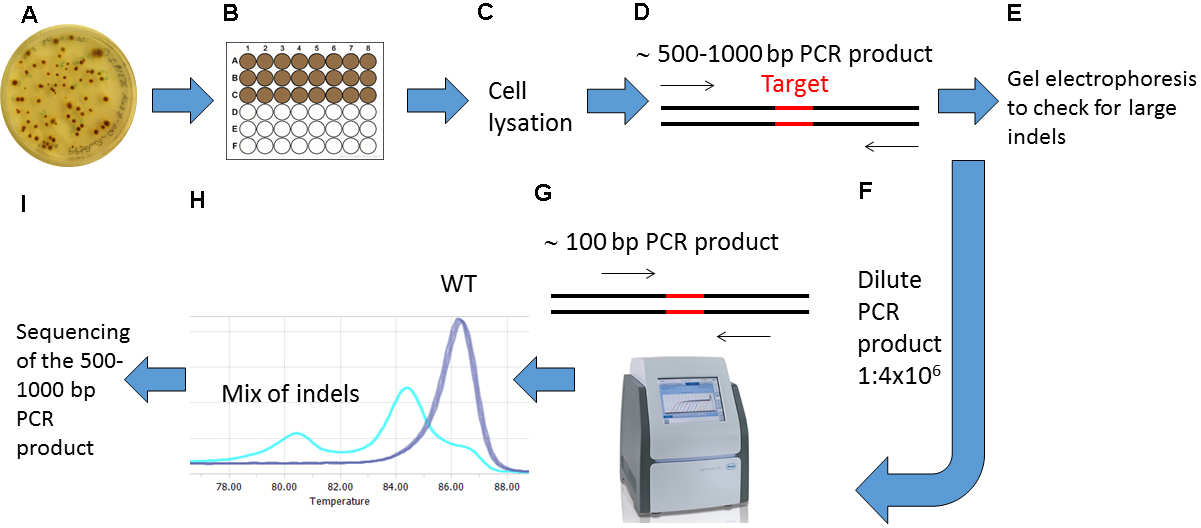
Figure 3. Overview of the screening procedure for detecting CRISPR/Cas9 induced mutations. A. Wait 2-4 weeks after biolistic transformation of P. tricornutum cells with the pKS diaCas9-sgRNA plasmid for colonies to appear on selection plates; B. Transfer the desired number of putative mutant colonies to a 24 or 48-well plate. Grow the cells for 1-2 weeks. C. Remove a small amount of cells from each well and lysate the cells; D. Use the lysate as template and amplify a 500-1,000 bp region around the target site by PCR; E. Check the PCR products by gel electrophoresis to identify large indels; F. Dilute the PCR product 1:4 x 106; G. Perform HRM analyses to detect smaller indels. Use the diluted PCR product from (F) as template in a PCR reaction that amplifies a region of around 100 bp surrounding the target site. H. Analyze the HRM data. Identify samples with CRISPR/Cas9 induced mutations by studying the difference in melting temperature between mutants and WT samples. I. Confirm mutations detected by HRM analyses by sequencing.- Preparations for High Resolution Melting (HRM) Analyses
- Pick transformed colonies and transfer them to 24 or 48-cell multiwell cell culture plates (VWR) containing f/2 medium with 50 µg/ml zeocin.
- After approx. 1-2 weeks of growth, transfer a small amount of cells (100-200 µl) to a 1.5 ml tube.
Note: Include a P. tricornutum wild type (WT) sample in addition to the transformed cells to be used as a control during the HRM analyses. - Centrifuge for 1 min at 17,000 x g, and remove the supernatant.
- Add 20 µl lysis buffer (see Recipes) and vortex for 30 sec.
- Place the tubes on ice for 15 min.
- Transfer the tubes to a heating block, and incubate for 10 min at 85 °C.
- Dilute the lysate 1:5 with DNase-free water, and centrifuge briefly to pellet cell debris. The diluted lysate can be used directly as template for PCR or stored at -20 °C.
- Amplify by PCR an amplicon with a size of approx. 500-1,000 bp surrounding the target of interest using the lysate from step D1g as template.
Note: A DNA polymerase that adds single deoxyadenosine (A) to the 3’ ends of the PCR products should be used to enable subsequent TOPO® TA Cloning (Invitrogen) of PCR products. - Check the PCR products by agarose gel electrophoresis. Transformants with large indels can be detected directly. HRM based PCR assays can be used to detect smaller indels (including single nucleotide indels).
- Pick transformed colonies and transfer them to 24 or 48-cell multiwell cell culture plates (VWR) containing f/2 medium with 50 µg/ml zeocin.
- HRM analyses
- Design HRM primers enabling amplification of a PCR product of approx. 100 bp surrounding the target site. The HRM primers should have annealing temperatures around 60 °C. Order HPLC purified primers.
- Make dilution series of the PCR products from step D1h from 1:40 to 1:4,000,000.
- Use 5 µl of the 1:4,000,000 diluted PCR product as template in a 20 µl PCR reaction using the LightCycler® 480 High Resolution Melting Master kit (Roche), with 1x LightCycler® 480 High Resolution Melting Master Mix, 200 nM forward and reverse primers and 3.0 mM MgCl2, final concentrations. Amplify the ~100 bp PCR product surrounding the target site in a LightCycler® 96 instrument (Roche), using (for most target sequences) the following cycling conditions: pre-incubation 10 min 95 °C, 45 cycles 3 step amplification (95 °C for 10 sec, 55 °C for 10 sec, 72 °C for 20 sec) followed by one cycle high resolution melting (95 °C for 60 sec, 40 °C for 60 sec, 65 °C for 1 sec, and finally using ramp rate 0.07 °C/sec and acquisition mode setting 15 readings/sec to reach target temperature 95 °C [1 sec]). Include 2-3 technical replicates for each sample.
- Analyze the raw data with the LightCycler® 96 software 1.1 (Roche) that sort the samples into groups based on normalized melting curves (Figure 4). Samples that show melting peaks that differ from WT are likely to contain PCR products with indels as a result of targeted genome editing induced by the CRISPR/Cas9 system.
- Confirm mutations detected by the HRM analysis by Sanger sequencing.
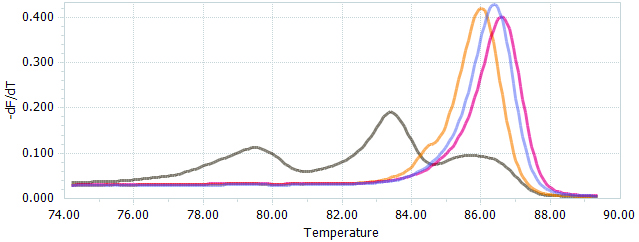
Figure 4. HRM data presented as normalized melting peaks. Purple line: profile of a WT PCR product. Red line: profile of a PCR product containing a single biallelic G-insert. Yellow line: profile of a PCR product containing a single biallelic A-insert. Brown line: profile of a mix of PCR products containing a variety of deletions of different sizes.
- Design HRM primers enabling amplification of a PCR product of approx. 100 bp surrounding the target site. The HRM primers should have annealing temperatures around 60 °C. Order HPLC purified primers.
- Preparations for High Resolution Melting (HRM) Analyses
- Identification of mutant lines originating from single cells with biallelic mutations
Note: The primary P. tricornutum colonies/cultures identified by HRM to contain targeted mutations are often a mix of cells with different indels. Some colonies also contain a low percentage of WT cells. Single cells must therefore be isolated from the primary cultures to obtain ‘clean’ mutant lines with biallelic mutations.- Spread 500 µl of a highly diluted cell suspension (~200 cells/ml) containing mutated P. tricornutum cells on f/2-Si, 1% (w/v) agar plates, 100 µg/ml Zeocin.
- Repeat the screening process (described in step D1) on the resulting colonies.
- Perform TOPO® TA Cloning (Invitrogen) to insert the 500-1,000 bp PCR product into a TOPO® TA-vector (e.g., pCRTM4-TOPO® TA vector [Invitrogen]) to identify the mutagenic event in the two different alleles. Direct Sanger sequencing of the PCR product will produce double peaks after the Cas9 target site if the two alleles contain different indels.
- Spread 500 µl of a highly diluted cell suspension (~200 cells/ml) containing mutated P. tricornutum cells on f/2-Si, 1% (w/v) agar plates, 100 µg/ml Zeocin.
Data analysis
HRM analysis is a post-PCR method that can be performed using the LightCycler® 96 software 1.1 (Roche) enabling easy screening of genetic variations in the PCR amplicons based on differences in the melting points of the amplicons. When performing HRM analysis, at least two technical replicates should be included per sample. Before the HRM analysis, the amplification curves and the melting curves from qPCR raw data should be evaluated. Amplification curves with high crossing point (Cp) values may contain unspecific PCR products, and curves giving > 30 Cp units should be excluded. The cycling conditions need to be optimized if the melting curves indicate unspecific PCR products in the WT samples or in the Non-template controls (NTCs). The crossing point between samples should not vary more than 5 Cp units. Mutations indicated by the HRM analyses must be confirmed by sequencing.
Recipes
- Lysis buffer
1% (v/v) Triton X-100
20 mM Tris-HCl pH 8
2 mM EDTA - LB medium/agar (per litre)
10 g pancreatic peptone
5 g Bacto yeast extract
5 g NaCl
15 g bacteriological agar (only for LB agar plates) - f/2 growth medium with natural (or artificial) seawater (Guillard’s medium for diatoms)
Filter seawater through a 0.2 µm sterile filter and autoclave at 120 °C for 20 min. Add f/2 trace elements, Vitamins and inorganic nutrients (see below) aseptically after autoclaving
Note: Filtering is necessary for the removal of inorganic and organic suspended particles from natural seawater, whereas autoclaving inactivates organisms small enough to pass through the pore size of the filter.
Stocks:- Trace elements (per litre)
4.36 g NA2EDTA
3.15 g FeCl2·6H2O
0.01 g CuSO4·5H2O
0.022 g ZnSO4·7H2O
0.01 g CoCl2·6H2O
0.18 g MnCl2·4H2O
0.006 g Na2Mo4·2H2O
Sterilize by filtering through a 0.2 µm sterile filter. Store in refrigerator (or freezer for long-term storage) - Vitamin mix (per litre)
0.0005 g cyanocobalamin (Vitamin B12)
0.1 g thiamine HCl (Vitamin B1)
0.0005 g biotin
Sterilize by filtering through a 0.2 µm sterile filter. Store in refrigerator (or freezer for long-term storage) - 30.0 g/L Na2SiO3·9H2O
Sterilize by autoclaving. Store in refrigerator - 75 g/L NaNO3
Sterilize by autoclaving. Store in refrigerator - 5.65 g/L NaH2PO4·2H2O
Sterilize by autoclaving. Store in refrigerator.
- 1 ml trace elements stock solution (1)
- 1 ml Vitamin mix stock solution (2)
- 1 ml Na2SiO3·9H2O stock solution (3)
- 1 ml NaNO3 stock solution (4)
- 1 ml NaH2PO4·2H2O stock solution (5)
- Trace elements (per litre)
Acknowledgments
This work was funded by a grant from the Gordon and Betty Moore Foundation GBMF 4966 to Atle Bones and the NTNU enabling technologies program.
References
- Falciatore, A., Casotti, R., Leblanc, C., Abrescia, C. and Bowler, C. (1999). Transformation of nonselectable reporter genes in marine diatoms. Mar Biotechnol (NY) 1(3): 239-251.
- Guillard, R. R. L. (1975). Culture of phytoplankton for feeding marine invertebrates. In: Smith, W. L. and Chanley, M. H. (Eds.). Culture of marine invertebrate animals: Proceedings–1st conference on culture of marine invertebrate animals Greenport. Springer 29-60.
- Hopes, A., Nekrasov, V., Kamoun, S. and Mock, T. (2016). Editing of the urease gene by CRISPR-Cas in the diatom Thalassiosira pseudonana. Plant Methods 12: 49.
- Nymark, M., Sharma, A. K., Sparstad, T., Bones, A. M. and Winge, P. (2016). A CRISPR/Cas9 system adapted for gene editing in marine algae. Sci Rep 6: 24951.
- Rastogi, A., Murik, O., Bowler, C. and Tirichine, L. (2016). PhytoCRISP-Ex: a web-based and stand-alone application to find specific target sequences for CRISPR/CAS editing. BMC Bioinformatics 17(1): 261.
- Sander, J. D. and Joung, J. K. (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32(4): 347-355.
- Shin, S. E., Lim, J. M., Koh, H. G., Kim, E. K., Kang, N. K., Jeon, S., Kwon, S., Shin, W. S., Lee, B., Hwangbo, K., Kim, J., Ye, S. H., Yun, J. Y., Seo, H., Oh, H. M., Kim, K. J., Kim, J. S., Jeong, W. J., Chang, Y. K. and Jeong, B. R. (2016). CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci Rep 6: 27810.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nymark, M., Sharma, A. K., Hafskjold, M. C. G., Sparstad, T., Bones, A. M. and Winge, P. (2017). CRISPR/Cas9 Gene Editing in the Marine Diatom Phaeodactylum tricornutum. Bio-protocol 7(15): e2442. DOI: 10.21769/BioProtoc.2442.
Category
Microbiology > Microbial genetics > Mutagenesis
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.