- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Demonstration and Quantification of Neutrophil Extracellular Trap Formation

Published: Vol 7, Iss 13, Jul 5, 2017 DOI: 10.21769/BioProtoc.2386 Views: 17457
Reviewed by: Benoit StijlemansMeenal SinhaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
In the recent decade, neutrophil extracellular traps (NETs) have been identified and confirmed as a new anti-microbial weapon of neutrophils. In this protocol, we describe easy methods to demonstrate NET formation by immunofluorescence staining of extracellular chromatin fiber with anti-DNA/Histone H1 antibody and quantification of NETs by using a non-cell-permeable DNA specific dye Sytox orange.
Keywords: NETs (neutrophil extracellular traps)Background
Neutrophils constitute the largest, evolutionary conserved fraction of circulating leukocytes. They set up the first defence line against pathogens by various mechanisms including the formation of neutrophil extracellular traps (NETs). During this process, activated neutrophils expel chromatin fibers from the nucleus. Invading pathogens are then trapped within the network of chromatin and killed by highly concentrated, NET-entangled antimicrobial proteins, such as myeloperoxidase (MPO) and elastase (Brinkmann et al., 2004). However, NETs are a double-edged sword; the unrestrained NET formation from over-activated neutrophils can also contribute to severe tissue damage, for instance by the cytotoxic effect of histone components of NETs (Saffarzadeh et al., 2012). One example of a pathological condition in which neutrophils are over-activated and have enhanced capacity to form NETs is systemic lupus erythematosus. The levels of antibodies against double-stranded DNA as well as other components of NETs are elevated in sera of lupus patients (Knight and Kaplan, 2012; Yu and Su, 2013). Affected skin and kidneys from lupus patients are infiltrated by netting neutrophils, which cause endothelial cell damage, a critical step in the pathogenesis of lupus and other neutrophil over-activation syndromes (Villanueva et al., 2011).
Different methods have been used for NET detection and quantification, including immunocytochemistry (Brinkmann et al., 2010), fluorescent dyes, flow cytometry, and ELISA. Immunocytochemistry with DAPI and DNA/histone was the best method for NET qualification and quantification, since decondensation of the nucleus indicates NET formation. Picogreen dye is a sensitive method to quantify NET-DNA concentration (Saffarzadeh et al., 2012; Tanaka et al., 2015), while Sytox orange is a fast and easy method for NET quantification. Flow cytometric analysis by measuring the signal for labeled-DNA/histone antibody, or myeloperoxidase-DNA based ELISA are useful methods for detection and quantification of NETs in pathological samples such as serum or peritoneal fluid of patients (Caudrillier et al., 2012).
In this protocol, we provide the details for the demonstration of NET formation by fluorescent immunostaining for chromatin fiber with anti-DNA/histone H1 antibody, which has a very high affinity for decondensed chromatin in NETs in comparison to DAPI or Hoechst (Saffarzadeh et al., 2012). Furthermore, we describe a quick method for NET quantification with Sytox orange, a non-cell-permeable DNA specific dye staining extracellular DNA content (Williams et al., 1999; Yost et al., 2009), by fluorescence intensity measured by a microplate reader. This protocol has been applied successfully in our recent studies, whereby we show that antibody- or complement-induced phagocytosis triggers rapid NET formation (Saffarzadeh et al., 2014), and more importantly, mesenchymal stem cells suppress NET formation from over-activated neutrophils (Jiang et al., 2016).
Materials and Reagents
- Pipette tips (Greiner Bio One International, Ultratip)
- Tube 50 ml (SARSTEDT, catalog number: 62.547.254 )
- Syringe 10 ml (B. Braun medical, catalog number: 4606108V-02 )
- Needle 26 G x ½” (B. Braun medical, catalog number: 4665457-02 )
- Pre-Separation filters (30 µm) (Miltenyi Biotec, catalog number: 130-041-407 )
- Millicell EZ Slide 8-well glass (EMD Millipore, catalog number: PEZGS0816 )
- 96-well, black, flat bottom plate, sterile, with lid (Corning, catalog number: 3916 )
- C57BL/6J mice at preferred age of 8-12 weeks, both male and female are suitable for this protocol (THE JACKSON LABORATORY, catalog number: 000664 )
- Phosphate buffered saline (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14190144 )
- 0.5 M EDTA, sterile (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15575020 )
- Histopaque-1077 (Sigma-Aldrich, catalog number: 10771 )
- Histopaque-1119 (Sigma-Aldrich, catalog number: 11191 )
- Optional: FITC-conjugated mouse anti-human CD15 antibody (Clone VIMC6) (Miltenyi Biotec, catalog number: 130-081-101 )
- Optional: FITC-conjugated mouse IgM isotype control (Miltenyi Biotec, catalog number: 130-093-178 )
- Optional: MACSxpress neutrophil isolation kit, human (Miltenyi Biotec, catalog number: 130-104-434 )
- Optional: MACSxpress erythrocyte depletion kit, human (Miltenyi Biotec, catalog number: 130-098-196 )
- Mouse neutrophil isolation kit (Miltenyi Biotec, catalog number: 130-097-658 )
- Optional: Propidium iodide (PI) staining solution (BD, BD Biosciences, catalog number: 556463 )
- Phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, catalog number: P8139 )
Note: Dissolve in DMSO to make 1 mg/ml stock, and store the aliquots at -20 °C. - Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D8418 )
- 16% (w/v) paraformaldehyde (PFA), methanol-free (Thermo Fisher Scientific, catalog number: 28908 )
- Purified mouse anti-DNA/Histone H1 antibody (EMD Millipore, catalog number: MAB3864 )
Note: Make aliquots and store at -20 °C. - 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Thermo Fisher Scientific, InvitrogenTM, catalog number: D1306 )
- Alexa Fluor 555-conjugated goat anti-mouse IgG secondary antibody (Thermo Fisher Scientific, InvitrogenTM, catalog number: A-21422 )
- Optional: APC-conjugated rat anti-mouse Ly6G (Gr-1) antibody (Clone RB6-8C5) (Thermo Fisher Scientific, eBioscienceTM, catalog number: 17-5931-82 )
- Optional: APC-conjugated rat IgG2b Isotype control (Clone eB149/10H5) (Thermo Fisher Scientific, eBioscienceTM, catalog number: 17-4031-82 )
- Optional: purified rat anti-mouse Ly6G antibody (Clone RB6-8C5) (Abcam, catalog number: ab25377 )
- Optional: purified rabbit anti-human CD15 antibody (Clone SP159) (Novus Biologicals, catalog number: NBP2-21754 )
- Purified goat anti-human/mouse myeloperoxidase/MPO antibody (R&D Systems, catalog number: AF3667 )
- Purified mouse IgG2a isotype control (Clone C1.18.4) (BD, BD Biosciences, catalog number: 550339 )
- Purified rabbit anti-human/mouse neutrophil elastase antibody (Abcam, catalog number: ab68672 )
- Sytox orange nucleic acid stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S11368 )
- Fetal bovine serum (FBS) (Biochrom, catalog number: S 0615 )
- Sodium azide (NaN3) (Sigma-Aldrich, catalog number: S2002 )
- RPMI 1640 medium (Thermo Fisher Scientific, GibcoTM, catalog number: 21875034 )
- GlutaMAX (Thermo Fisher Scientific, catalog number: 35050038 )
- MEM non-essential amino acids (NEAA) (Thermo Fisher Scientific, GibcoTM, catalog number: 11140035 )
- Penicillin/streptomycin (Biochrom, catalog number: A 2213 )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153 )
- Goat serum (Sigma-Aldrich, catalog number: G9023 )
- Fluorescence mounting medium (Agilent Technologies, DAKO, catalog number: S302380-2 )
- FACS buffer (can be kept at 4 °C for 2 weeks) (see Recipe 1)
- R1 medium (prepared media can be kept at 4 °C for 2 weeks) (see Recipe 2)
- Blocking buffer (prepare fresh) (see Recipe 3)
- Antibody diluent (prepare fresh) (see Recipe 4)
Equipment
- Pipettes
- Centrifuge
- Optional: MACSxpress Separator (Miltenyi Biotec, catalog number: 130-098-308 )
- Optional: MACSmixTM Tube Rotator (Miltenyi Biotec, catalog number: 130-090-753 )
- Humidified cell culture incubator set to 37 °C and 5% CO2
- Orbital shaker, such as Heidolph Unimax 1010 (Heidolph Instruments, model: Unimax 1010 , catalog number: 543-12310-00)
- Fluorescent microscope, such as Zeiss Axiophot microscope with an AxioCam digital color camera and AxioVision software v4.7 (Carl Zeiss, model: Axiophot )
- AxioCam digital color camera
- Microplate reader that can measure the absorbance and emission of Sytox orange at 547 nm and 570 nm, respectively, such as Mithras LB940 (BERTHOLD TECHNOLOGIES, model: Mithras LB 940 )
- Optional: Flow cytometer (to check the purity of isolated neutrophils) FACS Canto II (BD, BD Biosciences, model: BD FACSCANTO II ) with FACSDiva software
Software
- AxioVision software
- Optional: FACSDiva software
Procedure
- Neutrophil isolation
- Isolate human neutrophils from fresh peripheral blood using gradient centrifugation (Figures 1A and 1B)
- Dilute peripheral blood 1:2 with PBS containing 2 mM EDTA.
- In each 50 ml tube, carefully and sequentially lay 10 ml Histopaque-1119, 10 ml Histopaque-1077 and 25 ml diluted blood.
Note: Histopaque should be brought to room temperature prior to use. - Centrifugation at 700 x g (Note: Without break.) at room temperature for 30 min.
- Transfer the layer at the interface of Histopaque-1077 and Histopaque-1119 to a new 50 ml tube (the usual volume ranges from 2-3 ml), top up to 50 ml with PBS and centrifuge at 300 x g at room temperature for 10 min.
- Wash again with 30 ml PBS.
Note: The expected average yield of neutrophils per ml of peripheral blood from healthy donor ranges from 2-5 million cells.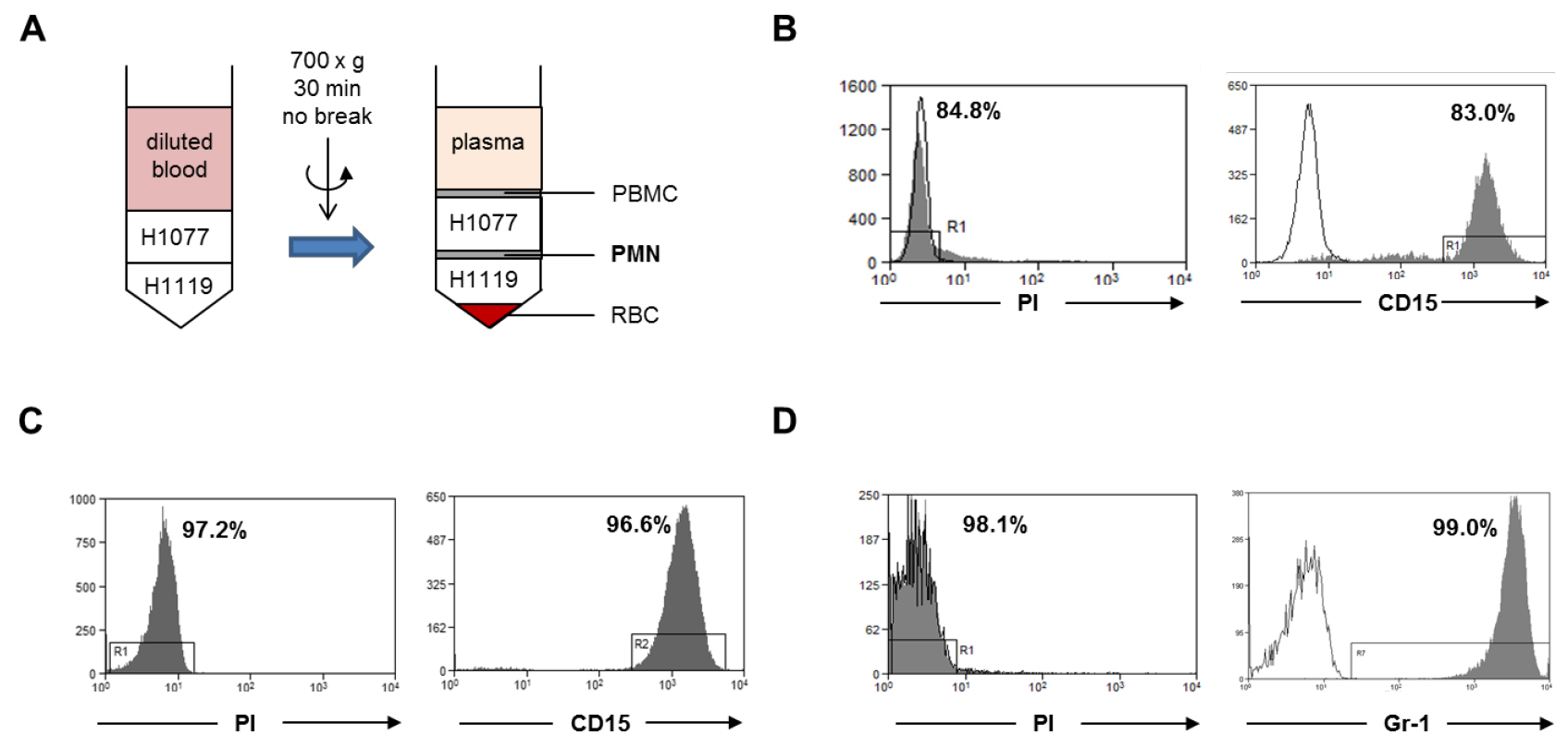
Figure 1. Neutrophil isolation. A. Human neutrophils were isolated from fresh peripheral blood of healthy donors by gradient centrifugation (step A1). The neutrophil layer at the interface of H1077 and H1119 were collected. PBMC, peripheral blood mononuclear cells; PMN, polymorphonuclear leukocytes; RBC, red blood cells. B. The cell viability was analyzed with PI dye and the purity of the viable neutrophils was analyzed using a human CD15 antibody within the PI negative gate. C. Human neutrophils were isolated from buffy coat with the MACSxpress method (step A2). The cell viability was analyzed with PI dye and the purity of the viable neutrophils was analyzed with human CD15 antibody within the PI negative gate. D. Murine bone marrow neutrophils were isolated by magnetic sorting using a negative depletion method (step A3). The cell viability was analyzed with PI dye and the purity of the viable neutrophils was analyzed with a mouse Ly6G (Gr-1) antibody within the PI negative gate. Gray histograms: PI dye or CD15 antibody or Gr-1 antibody; open histogram: unstained control or isotype antibody.
- Dilute peripheral blood 1:2 with PBS containing 2 mM EDTA.
- Alternative: Isolate human neutrophils with MACSxpress neutrophil isolation kit (Figure 1C)
- Mix 5 ml of buffy coat (Note: less than 12 h after collection.) with 1 ml reconstituted buffer A (kit component), 1 ml buffer B and 100 µl reconstituted MACSxpress erythrocyte depletion reagent.
- Incubate sample for 5 min at room temperature using the MACSmix Tube Rotator at 12 rpm.
- Place the open tube in the magnetic field of the MACSxpress Separator for 10 min, then carefully collect the supernatant containing neutrophils into a new tube.
- Mix 5 ml of buffy coat (Note: less than 12 h after collection.) with 1 ml reconstituted buffer A (kit component), 1 ml buffer B and 100 µl reconstituted MACSxpress erythrocyte depletion reagent.
- Isolation of murine bone marrow neutrophils (Figure 1D)
- Dissect femur bones of C57BL/6J mice, and flush out the bone marrow with 10 ml PBS containing 2 mM EDTA by using a 10 ml syringe and 26 G needle aseptically.
- Filter the suspension with a 30 µm cell strainer.
- Isolate neutrophils by immunomagnetic separation (MACS) using the negative depletion method with mouse neutrophil isolation kit (Miltenyi), following the manufacturer’s instructions.
Note: The expected average yield of neutrophils from bone marrow of two femur bones of one C57BL/6J mouse at age of 8-12 weeks ranges from 10-15 million cells.
- Dissect femur bones of C57BL/6J mice, and flush out the bone marrow with 10 ml PBS containing 2 mM EDTA by using a 10 ml syringe and 26 G needle aseptically.
- Optional: Check purity and viability of isolated neutrophils
- Resuspend 2 x 105 isolated human neutrophils in 100 µl FACS buffer (see Recipe 1) containing 5 µl of anti-human CD15-FITC antibody (or anti-mouse Gr1-APC antibody for mouse neutrophils). Another 2 x 105 cells in 100 µl FACS buffer containing 5 µl of mouse IgM-FITC isotype control (or rat IgG2b-APC isotype control for mouse neutrophils). Incubate at room temperature in the dark for 30 min.
- Add 100 µl FACS buffer, mix gently, and centrifuge to collect cell pellet with a benchtop centrifuge at 3,000 RPM (870 x g with 87 mm rotor) at room temperature, remove supernatant.
- Resuspend pellet in 300 µl FACS buffer, add 3 µl Propidium iodide (PI) staining solution, mix quickly.
- Proceed to flow cytometric analysis for CD15+PI- cells (human), or Gr-1hiPI- cells (mouse).
- Resuspend 2 x 105 isolated human neutrophils in 100 µl FACS buffer (see Recipe 1) containing 5 µl of anti-human CD15-FITC antibody (or anti-mouse Gr1-APC antibody for mouse neutrophils). Another 2 x 105 cells in 100 µl FACS buffer containing 5 µl of mouse IgM-FITC isotype control (or rat IgG2b-APC isotype control for mouse neutrophils). Incubate at room temperature in the dark for 30 min.
- Isolate human neutrophils from fresh peripheral blood using gradient centrifugation (Figures 1A and 1B)
- NET demonstration by immunostaining
Note: If the aim is to study the interaction of specific adherent cells with neutrophils and NET formation, seed the cells of interest one day prior to neutrophil isolation into 8-well glass chamber slide (Millicell EZ Slide 8-well glass) in 300 µl medium per well and culture overnight. The seeding cell number depends on the ratio of the target cells to neutrophils. Make duplicate wells for each condition. Remove culture medium before adding neutrophils.- Induction of NET formation
- Add freshly isolated neutrophils into 8-well glass chamber slide at a density of 2 x 105 neutrophils in 300 µl R1 medium (see Recipe 2) per well. Make duplicate wells for each condition.
Note: Resuspend neutrophils in R1 medium at 6.67 x 105 cells/ml, then add 300 µl per well. - Add the treatment of interest (e.g., potential chemical activator/inhibitor of NET, bacteria, interacting cells, etc.)
Note: Incubation time needs to be optimized individually, more details see Note 1. - Dissolve PMA in DMSO to make 1 mg/ml stock solution. To the positive control wells, and conditions that NET formation are desired, add PMA at a final concentration of 100 ng/ml to activate neutrophils (i.e., dilute PMA stock 1:100 in R1 medium, then add 3 µl of diluted PMA to each well). (see Note 2)
- Dilute DMSO 1:100 in R1 medium, add 3 µl of diluted DMSO to each well of the negative control (resting neutrophils) wells.
- Incubate for 3 h in a 37 °C incubator.
- Add 100 µl 4% PFA to each well without removing the medium (i.e., at final concentration of 1%) to fix the cells at 4 °C for overnight.
- Add freshly isolated neutrophils into 8-well glass chamber slide at a density of 2 x 105 neutrophils in 300 µl R1 medium (see Recipe 2) per well. Make duplicate wells for each condition.
- Staining with anti-DNA/Histone H1 antibody and DAPI on the next day (Figures 2A and 2B).
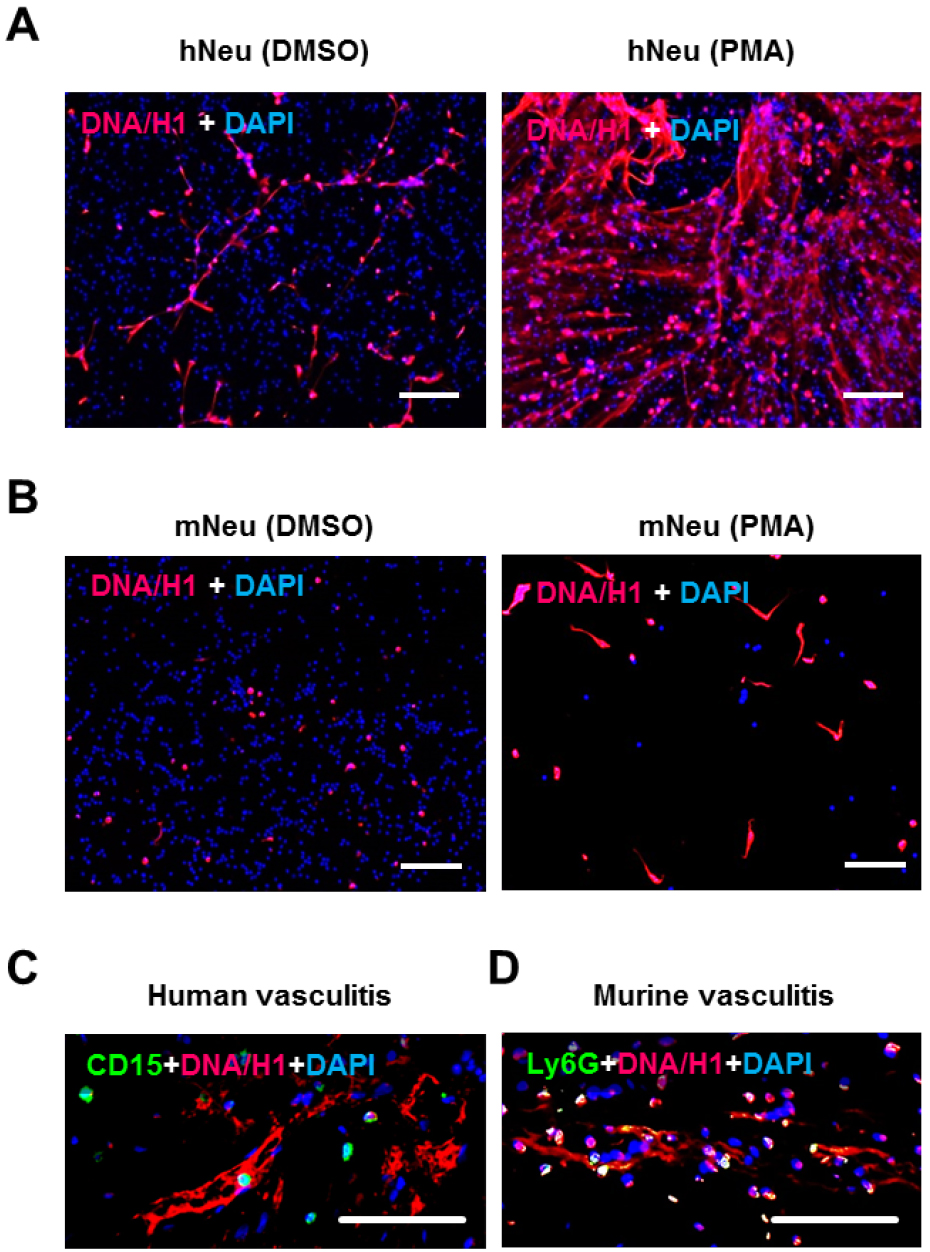
Figure 2. NET demonstration by immunostaining. 2 x 105 human neutrophils (A) or murine neutrophils (B) were activated by 100 ng/ml PMA for 3 h in wells of glass chamber slide. Resting neutrophils with DMSO served as negative control. Slides were fixed and labelled with anti-DNA/Histone H1 antibody followed by AF555-conjugated anti-mouse IgG (red) and nuclei were counterstained with DAPI (blue) (steps B1 and B2). C. Immunostaining of human neutrophil marker CD15 (green) and NET marker DNA/Histone H1 (red) on paraffin-embedded skin section from human vasculitis patients. D. Immunostaining of the murine neutrophil marker Ly6G (green) and NET marker DNA/Histone H1 (red) on paraffin-embedded skin section from mice with induced immune complex-mediated vasculitis (step B3). Scale bars = 100 µm.- Remove medium, add 100 µl PBS to each well and then remove.
- Add 100 µl blocking buffer (see Recipe 3) to each well. The slides were left still on bench at room temperature for 45 min.
- Dilute DNA/Histone H1 antibody in antibody diluent (see Recipe 4) to a final concentration of 4 µg/ml.
- Add 100 µl diluted antibody to each well, incubate on bench at room temperature for 1 h.
- Remove antibody, wash slides twice with 100 µl PBS in each well, on an orbital shaker at 100 RPM for 5 min for each time.
- Dilute secondary anti-mouse-AF555 1:200 in antibody diluent.
- Add 100 µl diluted secondary antibody to each well, incubate at room temperature in the dark for 45 min.
- Remove antibody, wash 2 times with 100 µl PBS on an orbital shaker.
- Dilute DAPI 5 mg/ml stock 1:5,000 in PBS, add 100 µl to each well, incubate at room temperature in the dark for 3 min.
- Remove DAPI staining solution, and remove the frame of chamber slides. Wash slides 2 times by dipping in a container with PBS.
- Mount cover slips with fluorescence mounting medium.
- Fluorescence microscopy with red channel (AF555) and blue channel (DAPI) (e.g., Zeiss Axiophot microscope with an AxioCam digital color camera and AxioVision software) (see Note 3).
- Remove medium, add 100 µl PBS to each well and then remove.
- Optional: staining with additional neutrophil marker (Figures 2C and 2D)
Additional neutrophil markers can be co-stained with NET, in order to provide an additional confirmation. The following primary antibodies have been tested, and one or more combinations can be considered depending on the number of color filters available on the fluorescence microscope: CD15 (human, Novus, 1:50), Ly6G (mouse, Abcam, 1:100), MPO (human and mouse, R&D Systems, 1:50), or NE (human and mouse, Abcam, 1:200). Proper secondary antibodies with different fluorophores other than NET staining are necessary. This staining can be performed before the NET staining. Sequential staining is recommended based on the staining specificity and intensity. - After blocking (step B2b), add 100 µl diluted primary antibody to each well, incubate at 4 °C overnight.
- Remove primary antibody, wash 2 times with 100 µl PBS.
- Add 100 µl diluted secondary antibody to each well, incubate at room temperature in the dark for 1 h.
- Remove secondary antibody, wash 2 times with 100 µl PBS.
- Continue with NET staining (step B2c).
- Induction of NET formation
- NET quantification with Sytox orange (Figure 3)
Note: If the aim is to study the interaction of specific adherent cells with neutrophils and NET formation, seed the cells of interest one day prior to neutrophil isolation into a 96-well, black, flat bottom plate in 100 µl medium per well, and culture overnight. The seeding cell number depends on the ratio of the target cells to neutrophils. Make 5 repeated wells for each condition. Remove culture medium before adding neutrophils.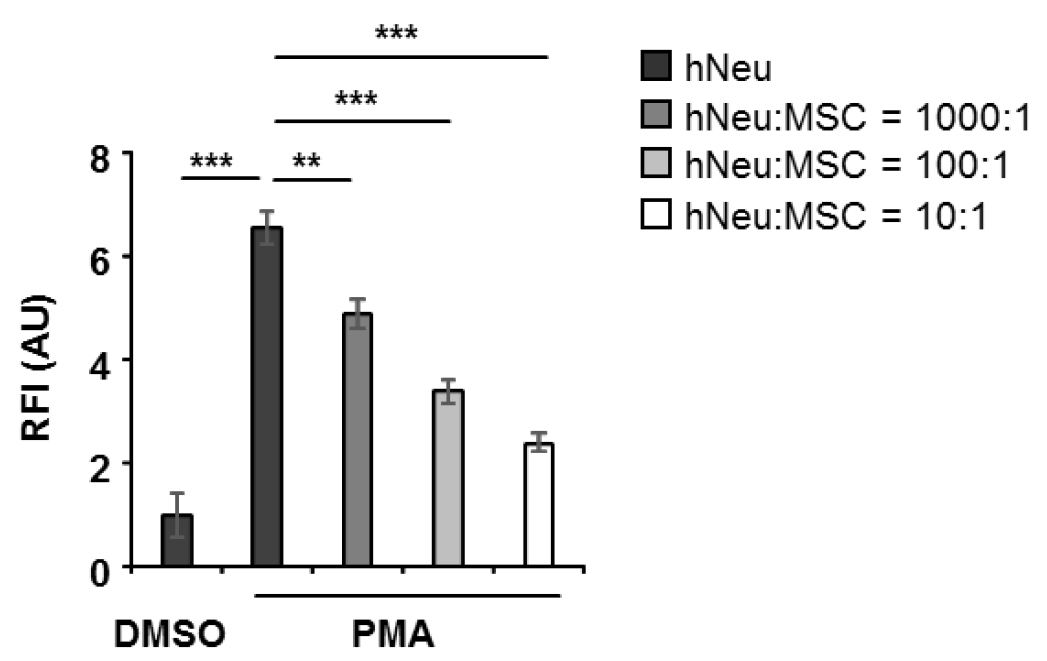
Figure 3. 1 x 105 human neutrophils (hNeu) were cultured alone or with adipose tissue derived mesenchymal stem cells (MSC) at hNeu:MSC ratios of 1,000:1, 100:1 and 10:1 and subsequently activated with 100 ng/ml PMA for 3 h. Cocultures were labelled with 0.25 µM Sytox orange for 5 min at room temperature and fluorescence intensities were measured by a microplate reader. Culture medium containing Sytox orange served as background fluorescent control. Fluorescence intensities were normalized on the intensity of non-activated controls. Results are expressed as mean ± SEM of quintuplicate measurements. This experiment was independently repeated three times with similar results. **P < 0.01; ***P < 0.001; RFI, relative fluorescence intensity; AU, arbitrary unit.- Add freshly isolated neutrophils into a 96-well, black, flat bottom plate at a density of 1 x 105 neutrophils in 100 µl R1 medium (see Recipe 2) per well. A background fluorescence control with only R1 medium, a positive control with PMA activation, and a negative DMSO-only control of resting neutrophil are necessary.
Note: Make 5 repeated wells for each condition. Avoid the wells in the first and last rows/columns of the plate. (We had experience that the peripheral wells of the plate may give inaccurate fluorescence reading, especially the wells on four corners. It could be due to the technical specification of the microplate reader or due to the quality of the black bottom 96-well plate.) - Add the treatment of interest (see Note 1 in Notes section).
- To the positive control wells, and conditions that NET formation are desired, add PMA at a final concentration at 100 ng/ml to activate neutrophils. (i.e., dilute 1 mg/ml PMA stock 1:100 in R1 medium, then add 1 µl of diluted PMA to each well).
- Dilute DMSO 1:100 in R1 medium, add 1 µl of diluted DMSO to each well of the negative control wells.
- Incubate for 3 h in a 37 °C incubator.
- Dilute 5 mM Sytox orange stock 1:5,000 in R1 medium. Then add 50 µl of 1 µM working solution to each well with a multichannel pipette (i.e., Sytox orange at a final concentration of 0.25 µM).
- Incubate for 5 min at room temperature in the dark.
- Centrifuge the plate at 400 x g for 5 min, carefully remove 100 µl supernatant from each well to reduce the background fluorescence.
Note: Remove the medium slowly and carefully, and avoid touching the bottom of the plate. - Immediately measure the fluorescence intensity with a Mithras LB940 microplate reader, at excitation and emission wavelengths of 540 nm and 580 nm, respectively (see Note 4).
- Add freshly isolated neutrophils into a 96-well, black, flat bottom plate at a density of 1 x 105 neutrophils in 100 µl R1 medium (see Recipe 2) per well. A background fluorescence control with only R1 medium, a positive control with PMA activation, and a negative DMSO-only control of resting neutrophil are necessary.
Data analysis
- Calculation of fluorescence intensity of Sytox orange assay
- Take the average of 5 repeats of R1 medium only condition. This is the background fluorescence of Sytox orange in medium.
- Subtract this background fluorescence from the fluorescence intensity readings of all other samples.
- Take the average of 5 repeats for each condition.
- Normalize the fluorescence intensity of each condition to the negative control (resting neutrophils), which is set as 1 arbitrary unit (AU).
- This assay should be performed at least 3 times with independent neutrophil samples. Data can be presented as mean ± SD (or SEM). Unpaired, two-tailed Student’s t-test can be used for significance test.
Notes
- The duration of treatment needs to be determined for each individual treatment condition. To study the effect of the target cells on NET formation, a proper incubation time should be considered to allow interaction of studying cells with neutrophils before adding the activator such as PMA. For example, to study the effect of mesenchymal stem cells (MSC) or human dermal fibroblasts (HDF) on NET formation, neutrophils are co-cultured with MSC or HDF for 2 h in R1 medium in a 37 °C incubator (Jiang et al., 2016).
- PMA is used to artificially trigger NET formation, for example, as the positive control. In conditions where the suppression of NET formation of the potential treatments is under investigation, the neutrophils are artificially activated with PMA too. However, if the potential treatments are expected to induce NET formation (e.g., bacterial), the addition of PMA is not necessary.
- The immunostaining pattern of NET looks different in human and mouse neutrophils. The NET fibers from human neutrophils are much longer and display more prominent network structure. Mouse bone marrow neutrophils are less capable of NET formation, but could be augmented by adding calcium to activate citrullination of histones which are important for NETosis. Regarding few nuclei in Figure 2B, based on our experience, compared to human neutrophils, smaller fraction of mouse neutrophils undergo NETosis. Oxidative burst and other activation processes lead to cell death and not adhering to the dishes.
- The microplate reader CLARIOstar with a monochromator from BMG Labtech (Ortenberg, Germany) has been tested for the Sytox orange assay, and it works nicely with excitation at 547 nm, and emission at 570 nm.
- The viability and purity of human neutrophils isolated from buffy coat depend on the freshness of the sample. Within 12 h of collection, the expected viability of neutrophils is higher than 80%. The expected purity from both gradient centrifugation and MACSxpress methods is higher than 80%. No difference is noticed in the functional response of neutrophils isolated by the two methods.
- In general, it is recommended to use different quantification methods to evaluate NETs, and to combine it with immunofluorescence microscopy to visualize NETs. The Sytox orange assay is useful as an easy NET quantification method for in vitro experiments. For in vivo conditions, a demonstration of neutrophil marker together with antibodies against NET components that can detect decondensed chromatin is recommended.
Recipes
- FACS buffer (can be kept at 4 °C for 2 weeks)
PBS containing 0.5% FBS, 0.02% NaN3, pH 7.4 - R1 medium (prepared media can be kept at 4 °C for 2 weeks)
RPMI1640 medium
1% FBS
1x GlutaMAX
1x MEM NEAA
1x penicillin/streptomycin - Blocking buffer (prepare fresh)
PBS containing 5% BSA together with 5% goat serum - Antibody diluent (prepare fresh)
PBS containing 1% BSA
Acknowledgments
This work was supported in part by research grants from the Baden-Württemberg Stiftung (P-BWS-ASII/15), the European Commission (CASCADE HEALTH-FP7-223236) and the German Research Foundation (SFB1149) to K.S.-K., the Baustein Program from the Medical Faculty, University of Ulm (LSBN.0100) to D.J., and from the Excellence Cluster Cardio-pulmonary System (ECCPS) to M.S. Part of figures are adapted and modified from the recent study of Jiang et al., 2016.
References
- Brinkmann, V., Laube, B., Abu Abed, U., Goosmann, C. and Zychlinsky, A. (2010). Neutrophil extracellular traps: how to generate and visualize them. J Vis Exp (36): 1724.
- Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., Weinrauch, Y. and Zychlinsky, A. (2004). Neutrophil extracellular traps kill bacteria. Science 303(5663): 1532-1535.
- Caudrillier, A., Kessenbrock, K., Gilliss, B. M., Nguyen, J. X., Marques, M. B., Monestier, M., Toy, P., Werb, Z. and Looney, M. R. (2012). Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 122(7): 2661-2671.
- Jiang, D., Muschhammer, J., Qi, Y., Kugler, A., de Vries, J. C., Saffarzadeh, M., Sindrilaru, A., Beken, S. V., Wlaschek, M., Kluth, M. A., Ganss, C., Frank, N. Y., Frank, M. H., Preissner, K. T. and Scharffetter-Kochanek, K. (2016). Suppression of neutrophil-mediated tissue damage-a novel skill of mesenchymal stem cells. Stem Cells 34(9): 2393-2406.
- Knight, J. S. and Kaplan, M. J. (2012). Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr Opin Rheumatol 24(5): 441-450.
- Saffarzadeh, M., Cabrera-Fuentes, H. A., Veit, F., Jiang, D., Scharffetter-Kochanek, K., Gille, C., Rooijakkers, S. H. M., Hartl, D. and Preissner, K. T. (2014). Characterization of rapid neutrophil extracellular trap formation and its cooperation with phagocytosis in human neutrophils. Discoveries 2(2): e19.
- Saffarzadeh, M., Juenemann, C., Queisser, M. A., Lochnit, G., Barreto, G., Galuska, S. P., Lohmeyer, J. and Preissner, K. T. (2012). Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 7(2): e32366.
- Tanaka, K., Okigami, M., Toiyama, Y., Okugawa, Y., Inoue, Y., Araki, T., Mohri, Y., Mizoguchi, A. and Kusunoki, M. (2015). Quantification of ex vivo neutrophil extracellular traps. Bio Protoc 5(15): e1549.
- Villanueva, E., Yalavarthi, S., Berthier, C. C., Hodgin, J. B., Khandpur, R., Lin, A. M., Rubin, C. J., Zhao, W., Olsen, S. H., Klinker, M., Shealy, D., Denny, M. F., Plumas, J., Chaperot, L., Kretzler, M., Bruce, A. T. and Kaplan, M. J. (2011). Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187(1): 538-552.
- Williams, M. A. and Solomkin, J. S. (1999). Integrin-mediated signaling in human neutrophil functioning. J Leukoc Biol 65(6): 725-736.
- Yost, C. C., Cody, M. J., Harris, E. S., Thornton, N. L., McInturff, A. M., Martinez, M. L., Chandler, N. B., Rodesch, C. K., Albertine, K. H., Petti, C. A., Weyrich, A. S. and Zimmerman, G. A. (2009). Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood 113(25): 6419-6427.
- Yu, Y. and Su, K. (2013). Neutrophil extracellular traps and systemic lupus erythematosus. J Clin Cell Immunol 4 (2): 139.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jiang, D., Saffarzadeh, M. and Scharffetter-Kochanek, K. (2017). In vitro Demonstration and Quantification of Neutrophil Extracellular Trap Formation. Bio-protocol 7(13): e2386. DOI: 10.21769/BioProtoc.2386.
Category
Immunology > Immune cell function > Neutrophil
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.