- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Targeted Nucleotide Substitution in Mammalian Cell by Target-AID

Published: Vol 7, Iss 11, Jun 5, 2017 DOI: 10.21769/BioProtoc.2339 Views: 19754
Reviewed by: Daan C. SwartsZhen ShiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Programmable RNA-guided nucleases based on CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated protein) systems have been applied to various type of cells as powerful genome editing tools. By using activation-induced cytidine deaminase (AID) in place of the nuclease activity of the CRISPR/Cas9 system, we have developed a genome editing tool for targeted nucleotide substitution (C to T or G to A) without donor DNA template (Figure 1; Nishida et al., 2016). Here we describe the detailed method for Target-AID to perform programmable point mutagenesis in the genome of mammalian cells. A specific method for targeting the hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene in Chinese Hamster Ovary (CHO) cell was described here as an example, while this method principally should be applicable to any gene of interest in a wide range of cell types.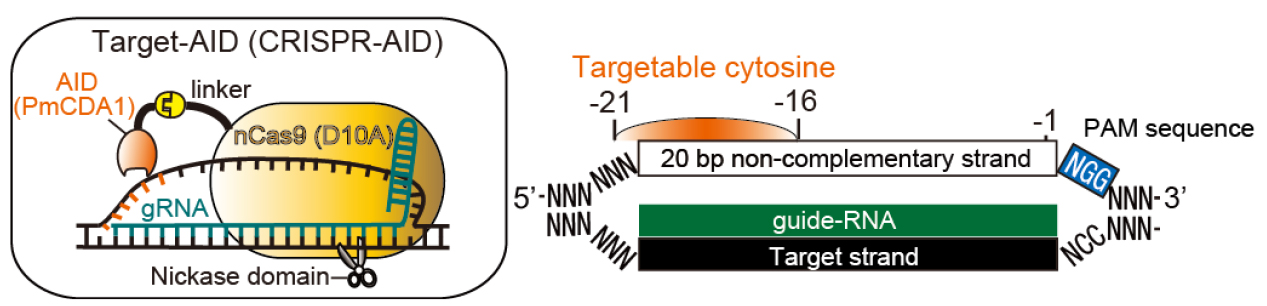
Figure 1. Schematic illustration for Target-AID and its targetable site. In a guide-RNA (gRNA)-dependent manner, PmCDA1 fused to nCas9 (D10A) via a linker performs programmable cytidine mutagenesis around -21 to -16 positions relative to PAM sequence on the non-complementary strand in mammalian cells. The targetable site was determined based on the efficient base substitution (> 20%) observed in the previous work.
Background
Insertion or deletion caused by DNA double strand break at the target site is efficiently induced to disrupt gene function. However, more precise genome modifications are still limited as homology directed repair is not always efficient enough in higher eukaryotes, especially when considering delivery of template DNA for in vivo genome editing. In addition, CRISPR nucleases also have some potential for off-target effect by cutting the genome (Cox et al., 2015). Target-AID demonstrated a very narrow range of targeted nucleotide modification without use of template DNA. AID can convert cytosine to uracil without DNA cleavage by deamination and then, uracil is converted to thymine or the other bases through DNA replication and/or repair. Use of uracil DNA-glycosylase inhibitor (UGI), which blocks removal of uracil in DNA and the subsequent repair pathway, rendered mutations more likely to be C to T substitutions and improved the efficiency. While a series of variable components for Target-AID had been tested such as linkage, nickase Cas9 (nCas9) and UGI in the original study, we will focus on the use of AID ortholog PmCDA1 derived from sea lamprey, fused to nCas9 or nCas9 plus UGI for simplicity. Consistent to our study, applying the rat apolipoprotein B mRNA editing enzyme, catalytic polypeptide (rAPOBEC1) has also been reported as a programmable base editor (BE). Although BE targeted 5 bases surrounding the -15 position upstream of PAM (Komor et al., 2016), Target-AID can modify 3 to 6 bases surrounding the -18 position upstream PAM. More recently, it has been reported that Target-AID can be applied for precise editing of plant genome (Shimatani et al., 2017).
Materials and Reagents
- Cell culture-treated polystyrene 24 well plate (Sumitomo Bakelite, catalog number: MS-80240Z )
- 100 mm dish (TPP, catalog number: 93100 )
- 15 ml and 1.5 ml tubes
- 200 μl pipette tips
- CHO-K1 cells (ECACC, catalog number: 85051005 )
- Target-AID vectors
nCas9-PmCDA1 (Addgene, catalog number: 79617 )
nCas9-PmCDA1-UGI (Addgene, catalog number: 79620 ) - Opti-MEM (Thermo Fisher Scientific, GibcoTM, catalog number: 31985070 )
- Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11668019 )
- Dulbecco’s phosphate buffered saline (D-PBS) (Nacalai Tesque, catalog number: 14249-24 )
- NucleoSpin Tissue XS (MACHEREY-NAGEL, catalog number: 740901.50 )
- A pair of primers to amplify the target genomic region plus 150-200 bp upstream and downstream sequences (for HPRT target1, Fw: 5’-GGCTACATAGAGGGATCCTGTGTCA-3’; Rev: 5’-ACAGTAGCTCTTCAGTCTGATAAAA-3’) (Eurofin genomics)
- KOD FX Neo (TOYOBO, catalog number: KFX-201 )
- Gel extraction kit (QIAGEN, catalog number: 28704 )
- (Optional) NEBNext Multiplex Oligos for Illumina (Dual Index Primer Set1) (New England Biolabs, catalog number: E7600S )
- (Optional) MiSeq reagent Kit v3 (Illumina, catalog number: MS-102-3003 )
- Ham’s F12 medium (Thermo Fisher Scientific, GibcoTM, catalog number: 11765054 )
- Fetal bovine serum (FBS) (Biosera, catalog number: FB-1360/500 )
- Penicillin-streptomycin (Nacalai Tesque, catalog number: 26253-84 )
- G418
- Trypsin-EDTA, 0.25% (Thermo Fisher Scientific, GibcoTM, catalog number: 25200056 )
- (Optional) 6-TG
- Ham’s F12 culture medium (see Recipes)
- Ham’s F12-G418 culture medium (see Recipes)
- Trypsin-EDTA 0.025% (see Recipes)
- (Optional) Ham’s F12-G418-6-TG culture medium (see Recipes)
Equipment
- Cell culture incubator at 37 °C with 5% CO2 (Panasonic, catalog number: KMCC17RU2J ) or equivalent
- Micropipette
- Optical microscope with 10x eyepiece and 10x objective lens (Olympus, model: CKX41 ) or equivalent
- Cell counting plate (WAKENBTECH, catalog number: OC-C-S02 )
- Centrifuge (Max speed: 15,000 rpm; Max RCF: 21,380 x g; 24 x 1.5/2.0 ml angle rotor; 12 x 15 ml swing-out rotor) (KUBOTA, model: 3740 ) or equivalent
- Heat block (TAITEC, model: CTU-Mini , catalog number: 0063288-000) or equivalent
- PCR thermal cycler (TaKaRa Bio, model: TP600 ) or equivalent
- Agarose gel electrophoresis system
- 3130xL Genetic Analyzer (Thermo Fisher Scientific, Applied BiosystemsTM, model: 3130xL Genetic Analyzer ) or equivalent
Software
- CLC Genomic workbench 7.0
Procedure
A schematic summary of Target-AID procedure described in this protocol can be found in Figure 2.
Figure 2. Schematic illustration for Target-AID vectors and experimental flow. The vector constructs were depicted on the top. The neomycin resistance gene (NeoR) is inserted downstream of 2A peptide to ensure the expression of the fusion protein in the G418-selected cells. The gRNA expression cassette can be replaced by digestion and ligation using ApaI and SpeI restriction enzyme sites. Experimental flow for targeting HPRT gene is depicted on the bottom. This experiment is divided into three procedures: (A) Transfection and selection; (B) Isolation of single-cell colony, and (C) Mutation analysis. Since HPRT converts a purine analog 6-thioguanine (6-TG) into a toxic derivative, HPRT gene disruption confers 6-TG resistance and can be counter-selected (HPRT gene mutation assay).
- Transfection of Target-AID plasmids into CHO-K1 cells
- Plate 0.5 x 105 CHO-K1 cells into a 24-well plate in 500 μl Ham’s F12 culture medium (see Recipes) for each well and culture at 37 °C with humidified 5% CO2 atmosphere for 24 h. The cells will be ~70-80% confluent at the time of transfection. Use appropriate medium and culture conditions for each cell type.
- Add 1.5 μg (in 2-5 μl solution) of each Target-AID plasmid to 50 μl Opti-MEM medium and mix gently.
- In a separate tube, add 3 μl Lipofectamine 2000 to 47 μl Opti-MEM medium per sample and mix gently.
- Add the Lipofectamine/Opti-MEM mixture (50 μl, from step A3) into each plasmid/Opti-MEM mixture (50 μl + plasmid; from step A2) and mix gently. Incubate at room temperature for 20 min.
- Remove the Ham’s F12 medium of the 24-well plate culture from step A1 and gently wash the adherent cells with 1x PBS. Repeat this wash process twice and add 500 μl Opti-MEM for each well.
- Using a micropipette, drop the plasmids-liposome solution (about 100 μl, from step A4) into each well to evenly distribute the solutions. For transient assay (optional), proceed to step A10.
- Incubate the transfected cells for 5 h at 37 °C with 5% CO2, and then replace the medium with Ham’s F12-G418 medium (see Recipes) after the wash procedure as described in step A5.
- Incubate the cells for 5-7 days at 37 °C with 5% CO2 condition. Replace the medium with fresh Ham’s F12-G418 every 3 days. For pulse incubation (optional), see step A9.
- (Optional) 24 h after transfection, transfer the culture plate to 25 °C with 5% CO2 condition and incubate for 24 h, followed by 37 °C incubation for 48 h. Repeat this process twice. This procedure is expected to increase mutation efficiency since PmCDA1 derived from sea lamprey is presumably adapted to lower temperatures (Nishida et al., 2016).
- (Optional) Incubate the transfected cells for 5 h at 37 °C with 5% CO2 and replace the medium with Ham’s F12 devoid of G418 medium. After 3 days, extract the genome from the cells and analyze the mutation efficiency by next generation sequencer (NGS) (see steps B5 and C1-C6). This analysis can detect the transient Target-AID expression mutagenesis.
- Plate 0.5 x 105 CHO-K1 cells into a 24-well plate in 500 μl Ham’s F12 culture medium (see Recipes) for each well and culture at 37 °C with humidified 5% CO2 atmosphere for 24 h. The cells will be ~70-80% confluent at the time of transfection. Use appropriate medium and culture conditions for each cell type.
- Picking up single-cell clones
- Five to seven days after transfection, wash the cells with 1x PBS and add 500 μl of 0.025% trypsin-EDTA (see Recipes) into each well. Incubate the plate for 2 min at 37 °C with 5% CO2.
- Pipette up and down thoroughly to remove the adherent cells, and transfer the trypsinized cells to a 1.5 ml tube. Add 500 μl Ham’s F12-G418 medium to stop the reaction.
- Centrifuge the cells at 1,500 x g for 1 min.
- Remove supernatant and resuspend the cell pellet in 100 μl Ham’s F12-G418 medium. Pipette up and down thoroughly to obtain single-cell suspension. Using a cell counting plate, check the single-cell suspension and count the cells. Adjust the final concentration to 1 x 103/ml.
- (Optional) The sample can be aliquoted for deep-sequencing analysis of the entire population as described by Nishida et al., 2016 (see step C6).
- Add 100 μl of single-cell suspension per 100 mm dish containing 7 ml Ham’s F12-G418 medium, and mix gently. Make replicates at least three times as needed for statistical analysis. The mutation efficiency will be analyzed by appropriate statistical analysis such as Student’s t-test. (Optional) For counter-selection of the HPRT-null cells, add 100-300 μl of single-cell suspension per 100 mm dish containing 7 ml Ham’s F12-G418-6-TG medium (see Recipes) and mix gently. Make plates with different concentration of cells, as survival rate may vary depending on the target. Efficiency for HPRT gene disruption will be estimated by the rate of 6-TG-resistant colonies over G418-resistant colonies. Incubate the plate at 37 °C with 5% CO2.
- After 5-10 days incubation, colonies should be visible on the dish.
- Remove the medium, then pick up single colonies using a 200 μl pipette tip and transfer the colony to a well of 24-well plate containing Ham’s F12 medium-G418. Single-cell pick-up can be performed by typical methods such as trypsinization and serial dilution method.
- Incubate at 37 °C with 5% CO2 for over a week until the clones to become 70-80% confluent in a 24-well plate.
- Five to seven days after transfection, wash the cells with 1x PBS and add 500 μl of 0.025% trypsin-EDTA (see Recipes) into each well. Incubate the plate for 2 min at 37 °C with 5% CO2.
- DNA sequencing analysis of the induced mutations
- Extract genomic DNA using NucleoSpin Tissue XS according to the manufacturer’s instructions.
- Perform PCR with primers (see ‘Materials and Reagents’) approximately 200 bp flanking each side of the target site using KOD FX Neo polymerase according to the manufacturer’s instructions.
- Check the PCR products by gel electrophoresis.
- Isolate and purify the products by using QIAGEN gel extraction kit according to the manufacturer’s instructions.
- Using 3130xL Genetic Analyzer, sequence the DNA and confirm the mutations induced by Target-AID (Figure 3).
- (Optional) To detect minor mutations and obtain comprehensive mutational spectrum, deep sequencing by NGS can be applied. By using extracted genomic DNA, perform a 1st PCR with a pair of primers flanking a region of approximately 500 bp with the target site at the center. Check and purify the PCR products (see steps C3-C4) on an agarose gel. Using the first PCR product as a template, perform a second nested PCR with primers containing an adaptor sequence (Fw, 5’-TCTTTCCCTACACCGACGCTCTTCCGATCT-(forward target specific sequence)-3’; Rev, 5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-(reverse target specific sequence)-3’), to amplify the adaptor added-amplicon (~300 bp) fragment including the target site at the center. Label the fragment with index sequences by using NEBNext Multiplex Oligos for Illumina sequencing according to the manufacturer’s instructions. Using the MiSeq system, perform deep-sequencing analysis to obtain paired 300 bp length and > 100,000 reads per sample on average, according to the manufacturer’s instructions.

Figure 3. Representative mutation alignment induced by Target-AID. Mutations induced by transfection with nCas9(D10A)-PmCDA1 (79619) vector contain not only nucleotide substitutions but also specific insertions and deletions. Mutations are more likely to be C to T substitutions when a vector expressing the nCas9-UGI fusion (79620) is used. Mutations including the insertion, deletion and point mutation were shown in red.
- Extract genomic DNA using NucleoSpin Tissue XS according to the manufacturer’s instructions.
Data analysis
Data analysis using CLC Genomic workbench 7.0 was described as ‘Deep sequencing of target and off-target region of CHO cells’ in Nishida et al., 2016.
Notes
- There are various methods for the single-cell cloning. The optimal protocol for each cell type should be used.
- By modifying the Cytosine at the antisense strand, targeted Guanine to Adenine substitutions can be introduced into the sense strand.
Recipes
- Ham’s F12 culture medium
Ham’s F12 supplemented with 10% FBS and 100 μg/ml penicillin-streptomycin - Ham’s F12-G418 culture medium
Ham’s F12 supplemented with 10% FBS, 100 μg/ml penicillin-streptomycin and 125 μg/ml G418 - Trypsin-EDTA 0.025%
Trypsin-EDTA 0.25% diluted 10-fold with D-PBS - (Optional) Ham’s F12-G418-6-TG culture medium
Ham’s F12 supplemented with 10% FBS, 100 μg/ml penicillin-streptomycin, 125 μg/ml G418 and 5 g/ml 6-TG
Acknowledgments
This protocol was originally published as part of Nishida et al., 2016. This work was supported by the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from Japan Agency for Medical Research and Development (AMED). This work was also partly supported by a Special Coordination Fund for Promoting Science and Technology, Creation of Innovative Centers for Advanced Interdisciplinary Research Areas (Innovative Bioproduction Kobe) from the Ministry of Education, Culture, Sports and Technology (MEXT) of Japan; Cross-ministerial Strategic Innovation Promotion Program; JSPS KAKENHI [Grant Number 26119710, 16K14654 and 15K18647]; the New Energy and Industrial Technology Development Organization (NEDO) and Cross-ministerial Strategic Innovation Promotion Program (SIP).
References
- Cox, D. B., Platt, R. J. and Zhang, F. (2015). Therapeutic genome editing: prospects and challenges. Nat Med 21(2): 121-131.
- Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533(7603): 420-424.
- Nishida, K., Arazoe, T., Yachie, N., Banno, S., Kakimoto, M., Tabata, M., Mochizuki, M., Miyabe, A., Araki, M., Hara, K. Y., Shimatani, Z. and Kondo, A. (2016). Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353 (6305).
- Shimatani, Z., Kashojiya, S., Takayama, M., Terada, R., Arazoe, T., Ishii, H., Teramura, H., Yamamoto, T., Komatsu, H., Miura, K., Ezura, H., Nishida, K., Ariizumi, T. and Kondo, A. (2017). Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat Biotechnol 35(5): 441-443.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Arazoe, T., Nishida, K. and Kondo, A. (2017). Targeted Nucleotide Substitution in Mammalian Cell by Target-AID. Bio-protocol 7(11): e2339. DOI: 10.21769/BioProtoc.2339.
Category
Cancer Biology > General technique > Genetics > DNA damage
Cell Biology > Cell engineering > CRISPR-cas9
Molecular Biology > DNA > DNA damage and repair
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.