- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Modification of 3’ Terminal Ends of DNA and RNA Using DNA Polymerase θ Terminal Transferase Activity
Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2330 Views: 10485
Reviewed by: Gal HaimovichVamseedhar RayaproluDavid Paul
Original research article
The authors used this protocol in:

Jun 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
DNA polymerase θ (Polθ) is a promiscuous enzyme that is essential for the error-prone DNA double-strand break (DSB) repair pathway called alternative end-joining (alt-EJ). During this form of DSB repair, Polθ performs terminal transferase activity at the 3’ termini of resected DSBs via templated and non-templated nucleotide addition cycles. Since human Polθ is able to modify the 3’ terminal ends of both DNA and RNA with a wide array of large and diverse ribonucleotide and deoxyribonucleotide analogs, its terminal transferase activity is more useful for biotechnology applications than terminal deoxynucleotidyl transferase (TdT). Here, we present in detail simple methods by which purified human Polθ is utilized to modify the 3’ terminal ends of RNA and DNA for various applications in biotechnology and biomedical research.
Keywords: DNA polymeraseBackground
The human POLQ gene encodes a large protein that contains an N-terminal superfamily 2 (SF2) type helicase domain and a C-terminal A-family polymerase domain (Sfeir and Symington, 2015; Black et al., 2016; Wood and Doublie, 2016). The protein also encodes for a large central domain whose function has yet to be ascribed. Polθ is expressed in metazoans and has been shown to function in multiple aspects of DNA replication and repair (Black et al., 2016; Wood and Doublie, 2016). Recent work showed that mammalian Polθ is essential for the error-prone DNA double-strand break (DSB) repair pathway called alternative end-joining (alt-EJ), also known as microhomology mediated end-joining (MMEJ) (Yousefzadeh et al., 2014; Kent et al., 2015; Mateos-Gomez et al., 2015). This essential function of the polymerase is conserved among metazoans (Chan et al., 2010; Koole et al., 2014).
Interestingly, Polθ mediated alt-EJ results in relatively large deletions and insertions (indels) at DNA repair junctions compared to the more accurate non-homologous end-joining (NHEJ) pathway (Black et al., 2016). For instance, alt-EJ typically generates insertions ranging from 1-6 base pairs (bp), and in some cases insertions can exceed 30 bp (Yousefzadeh et al., 2014; Mateos-Gomez et al., 2015; Black et al., 2016; Kent et al., 2016). Intriguingly, multiple studies from invertebrates and vertebrate systems show that some insertion tracts are templated by nearby DNA sequences such as those flanking the DSB (Black et al., 2016). In other cases, insertion sequences appear to be random (Black et al., 2016). These and other studies led to the idea that Polθ might generate insertion tracts at DSBs by both templated and non-templated terminal transferase mechanisms.
Indeed, in a recent study Kent et al. demonstrated that the human Polθ polymerase domain, hereinafter referred to as Polθ, exhibits robust terminal transferase activity preferentially on single-strand DNA (ssDNA) and double-strand DNA containing 3’ ssDNA overhangs, referred to as partial ssDNA (pssDNA) (Kent et al., 2016). This study also compared the terminal transferase activities of Polθ and terminal deoxynucleotidyl transferase (TdT) using their respective optimal conditions, and found that Polθ is a more versatile enzyme for modifying the 3’ terminus of nucleic acids. For example, the authors showed that Polθ is able to modify nucleic acids with a wider variety of nucleotide analogs, such as those containing large fluorophores or attachment chemistries (Kent et al., 2016). As a specific example, Polθ was shown to efficiently modify ssDNA with a nucleotide analog containing click chemistry applicability (i.e., a linker attached to an azide group), whereas TdT failed to use the same nucleotide as a substrate (Kent et al., 2016). TdT was also unable to use a Texas Red conjugated nucleotide analog that Polθ efficiently utilized to modify ssDNA (Kent et al., 2016). Polθ is also capable of modifying the 3’ terminal ends of RNA and appears to show a significantly lower discrimination against ribonucleotides compared to TdT (Kent et al., 2016). Altogether, this recent report demonstrates that Polθ is a more versatile terminal transferase enzyme than TdT and therefore should be more useful for a wide range of applications in biotechnology and biomedical research that require modification of 3’ terminal DNA and RNA ends (Kent et al., 2016). Here, we explain in detail step-by-step procedures for using Polθ as a robust terminal transferase enzyme in vitro.
Materials and Reagents
- The following reagents are needed for modifying nucleic acids with Polθ:
- Pipette tips (i.e., Fisher Scientific, catalog number: 02-707-432 )
- Microcentrifuge tubes (i.e., 0.5 ml or 1.5 ml). DNase and RNase free tubes are recommended for reactions (i.e., BioDot Ultra Spin 1.5 ml Microcentrifuge Tubes) (DOT Scientific, catalog number: 711-FTG )
- ssDNA or RNA to be modified (typically 10-50 nt in length; desalted, HPLC or PAGE purified)
- Purified Polθ (residues 1,792-2,590, MW = 90 kDa) (expression vector and purification methods: Hogg et al., 2011)
- Nucleoside triphosphate analogs (i.e., TriLink BioTechnologies, catalog number: N-2008-102502 and TriLink BioTechnologies, catalog number: N-5001 ) or canonical nucleoside triphosphates (i.e., Promega, catalog number: U120 )
- 1 M Tris buffer pH 8.2 (i.e., DOT Scientific, catalog number: DST60040-10000 )
- Hydrochloric acid (HCl) (i.e., Thomas Scientific, catalog number: C395L46 )
- NP-40 detergent (i.e., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 28324 )
- Bovine serum albumin (BSA) (protease free) (i.e., Fisher Scientific, catalog number: BP9703100 )
- Manganese(II) chloride tetrahydrate (MnCl2·4H2O) (i.e., Sigma-Aldrich, catalog number: M3634-100G )
- 1 M HEPES buffer pH 8.0 (i.e., Oakwood Products, catalog number: 047861-1Kg )
- Sodium hydroxide (NaOH)
- Deionized water (dH2O) (Autoclaved Nanopure filtered water is recommended for reactions with RNA)
- 1 M Tris buffer pH 8.2 (see Recipes)
- Buffer A (see Recipes)
- 1 M HEPES buffer pH 8.0 (see Recipes)
- Pipette tips (i.e., Fisher Scientific, catalog number: 02-707-432 )
- The following reagents are needed if visualization of RNA and DNA modification is desired:
- Ammonium persulfate (APS) (i.e., Sigma-Aldrich, catalog number: A3678 )
- 40% acrylamide solution (19:1 acrylamide:bis acrylamide) (i.e., Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9022 )
- Ethylenediaminetetraacetic acid (EDTA) (i.e., Sigma-Aldrich, catalog number: 03620 )
- Tetramethylethylenediamine (TEMED) (i.e., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 17919 )
- Formamide (i.e., Sigma-Aldrich)
- 1 M HEPES buffer pH 8.0 (i.e., Oakwood Products, catalog number: 047861-1Kg )
- Sodium chloride (NaCl) (i.e., Sigma-Aldrich, catalog number: S9888 )
- Glycerol (i.e., Avantor Performance Materials, Macron Fine Chemicals, catalog number: 5092-16 )
- NP-40 detergent (i.e., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 28324 )
- Xylene cyanol (i.e., Fischer Scientific, catalog number: BP125-100 )
- Bromophenol blue (i.e., DOT Scientific, catalog number: DSB40160-25 )
- Dithiothreitol (DTT) (i.e., bioWORLD, catalog number: 40400120 )
- Buffer B (see Recipes)
- 2x stop buffer (see Recipes)
- Ammonium persulfate (APS) (i.e., Sigma-Aldrich, catalog number: A3678 )
Equipment
- The following equipment is needed for modifying nucleic acids with Polθ:
- Pipettes (i.e., P2, P20)
- Temperature controlled water bath or incubator
- Pipettes (i.e., P2, P20)
- The following equipment is needed if visualization of RNA and DNA modification is desired:
- Large vertical sequencing gel apparatus (i.e., APOGEE ELECTROPHORESIS, model: Model S2 ) or small vertical gel apparatus (i.e., Bio-Rad Laboratories)
- Glass plates for large or small gels
Note: Large glass plates for APOGEE ELECTROPHORESIS Model S2 SEQUENCER can be obtained from APOGEE ELECTROPHORESIS, and standard small plates and apparatuses can be obtained from Bio-Rad Laboratories. - Plastic combs for gels
Note: Plastic combs for large sequencing gels can be obtained from APOGEE ELECTROPHORESIS. - Gel spacers 0.4 mm thick (i.e., APOGEE ELECTROPHORESIS)
- Electrophoresis power supply (standard voltage [i.e., 300 V] source for small gels, high voltage [i.e., 5,000 V] source for large sequencing gels)
Note: Power supplies may be obtained from Bio-Rad Laboratories. - Fluorescence imaging system (for fluorophore labeled nucleic acids)
- Film developer or phosphorimager (i.e., FUJIFILM, model: FLA7000 ) (for 5’ 32P radio-labeled nucleic acids)
- Large vertical sequencing gel apparatus (i.e., APOGEE ELECTROPHORESIS, model: Model S2 ) or small vertical gel apparatus (i.e., Bio-Rad Laboratories)
Procedure
- Modification of the 3’ terminal ends of DNA and RNA using Polθ
- Purified Polθ is needed for modifying the 3’ terminal ends of DNA and RNA. Procedures for expressing and purifying Polθ from E. coli are described in previous studies (Hogg et al., 2011).
Notes:
- Synthetic single-stranded DNA (ssDNA) and RNA typically ~10-50 nt (nucleotides) in length have been routinely modified in our laboratory. Thus, we recommend using nucleic acids of similar length for the following procedure.
- All reagents below are listed as final concentrations.
- A typical procedure for modifying ssDNA or RNA is described as follows:
- 50-100 nM of the ssDNA or RNA to be modified is mixed with 50-500 μM concentration of the desired nucleotide used for modification in buffer A (see Recipes) along with 5 mM MnCl2 in a reaction volume of 10-20 μl.
Note: We have not thoroughly tested the effects of different nucleotide concentrations. However, concentration ranges between 50-500 μM have shown efficient terminal transferase activity. Most of our previous reactions included 500 μM nucleotides. Reaction volumes can be varied according to preference and concentrations of ssDNA and RNA used successfully in the reaction in our experience are 50-100 nM. Importantly, the DNA or RNA must include a 3’ terminal nucleotide containing a hydroxyl group at the 3’ position of the sugar moiety for Polθ terminal transferase activity to occur. - The terminal transferase reaction is then initiated by adding 200-500 nM purified Polθ and incubating at 42 °C for 2 h. Reactions are gently mixed with a pipette. Vortexing is not recommended.
- Reactions can be terminated by heating to ≥ 80 °C for 10 min or by the addition of ≥ 10 mM EDTA. Although the amount of Polθ can vary, optimal terminal transferase activity is observed with a 5-10 higher ratio of polymerase to nucleic acid molecule. We note that relatively high concentrations (i.e., > 50 mM) of salt (e.g., NaCl) suppress Polθ terminal transferase activity.
- The 2 h incubation time specified in the above procedure will give rise to multiple (i.e., 3 to > 100) terminal transfer events for most canonical nucleotides. However, in some cases only a single transfer event may occur depending on the particular nucleotide analog used. For example, certain nucleotide analogs may not be efficiently incorporated by Polθ and thus limit the enzyme to a single nucleotide transfer event. For determining the number of terminal transferase events that occur on a given substrate in the presence of particular nucleotides, we recommend visualizing the initial nucleic acid substrate and nucleic acid reaction products in a denaturing sequencing gel as described below in the Data analysis section.
- Purified Polθ is needed for modifying the 3’ terminal ends of DNA and RNA. Procedures for expressing and purifying Polθ from E. coli are described in previous studies (Hogg et al., 2011).
- Examples of experimental procedures for modifying the 3’ terminal ends of DNA and RNA using Polθ
- As examples of Polθ terminal transferase activity on ssDNA and RNA, reactions were performed as follows. 50 nM of 5’ 32P radio-labeled ssDNA oligo (sequence indicated; Figures 1A and 1B) or RNA (sequence indicated; Figure 1C) was mixed with 50 µM (Figures 1A and 1B) or 500 µM.
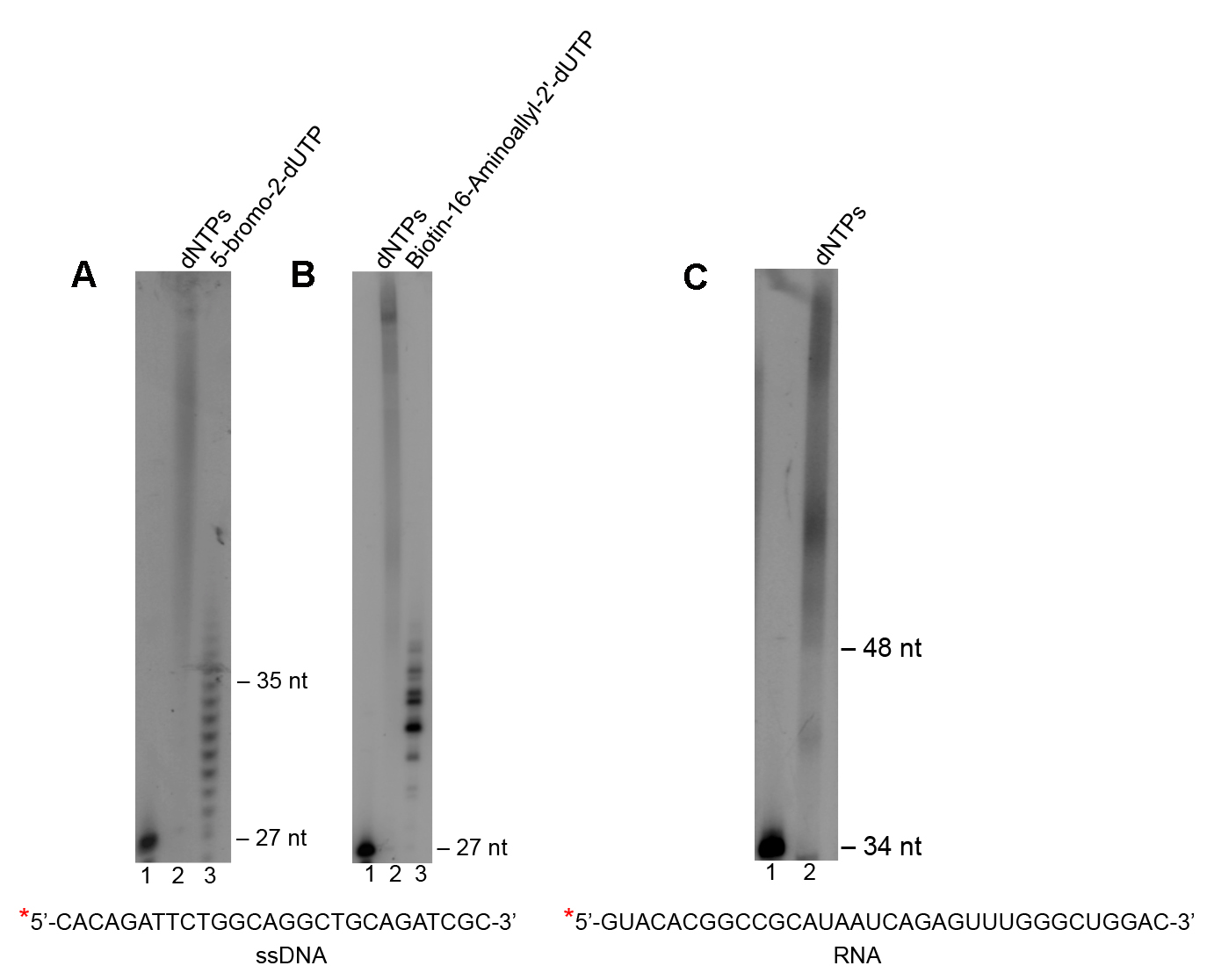
Figure 1. Use of Polθ terminal transferase activity to modify the 3’ terminal ends of DNA and RNA. A-C. Denaturing gels showing Polθ terminal transferase activity on the indicated ssDNA (A and B) and RNA (C) substrates in the presence of the indicated nucleotides. Lanes 1 lack Polθ and nucleotides. Lanes 2 and 3 include Polθ and the indicated nucleotides. ssDNA and RNA sequences are indicated at bottom. *, 32P radio-label. - (Figure 1C) of the indicated nucleotides along with 5 mM MnCl2 in 20 µl of buffer A (see Recipes).
- Reactions were initiated by adding 200 nM of purified Polθ (stored in buffer B [see Recipes]), then incubating at 42 °C.
- After 2 h, reactions were terminated by adding 20 µl of 2x stop buffer (see Recipes).
- Radio-labeled ssDNA and RNA were then analyzed after denaturing gel electrophoresis and autoradiography as described below in the Data analysis section. The data show that Polθ efficiently transfers nucleotides to the 3’ terminus of nucleic acid substrates as demonstrated in previous studies (Figure 1) (Kent et al., 2016). These experiments also demonstrate the ability of Polθ to efficiently transfer large nucleotide analogs, consistent with recent work (Kent et al., 2016). We note that Polθ may also be used to modify double-strand blunt ended DNA, however, fewer nucleotides are transferred to these substrates as demonstrated in previous work (Kent et al., 2016). Partial single-strand DNA containing 3’ overhangs are most efficiently extended by Polθ (Kent et al., 2016).
- As examples of Polθ terminal transferase activity on ssDNA and RNA, reactions were performed as follows. 50 nM of 5’ 32P radio-labeled ssDNA oligo (sequence indicated; Figures 1A and 1B) or RNA (sequence indicated; Figure 1C) was mixed with 50 µM (Figures 1A and 1B) or 500 µM.
Data analysis
- Visualizing modified RNA and DNA in denaturing gels
- Reactions should be performed as above with the following modifications. The RNA or DNA should be either 5’ radio-labeled, or conjugated with a fluorophore prior to the reaction for their detection in denaturing gels. Nucleic acids can be radio-labeled using T4 polynucleotide kinase in the presence of gamma-ATP. For fluorophore detection, DNA and RNA oligonucleotides can be purchased with 5’ fluorophore linkages. We recommend terminating reactions with an equal volume of 2x stop buffer (see Recipes).
- Initial nucleic acid substrates and reaction products should be resolved in standard urea denaturing 10-20% polyacrylamide gels. Helpful protocols for pouring and processing urea denaturing polyacrylamide sequencing gels are referenced here (Summer et al., 2009; Flett, et al., 2013). Large sequencing gels will allow for the highest resolution (i.e., single nucleotide resolution). However, smaller gels may provide enough resolution depending on the particular application. In the case of RNA, we recommend adding 10-15% formamide to urea denaturing polyacrylamide gels to reduce RNA secondary structures that can appear as smears in the gel. Large sequencing gels are typically run at 70-80 W using a high voltage (5,000 V) power supply. The resolved nucleic acids can then be visualized using a fluorescent imager (for 5’ fluorophore conjugated oligos) or using a phosphorimager or autoradiography (for 5’ 32P labeled oligos). Figure 1 shows examples of 5’ 32P radio-labeled nucleic acids that were resolved in large sequencing gels, then visualized by autoradiography.
- Reactions should be performed as above with the following modifications. The RNA or DNA should be either 5’ radio-labeled, or conjugated with a fluorophore prior to the reaction for their detection in denaturing gels. Nucleic acids can be radio-labeled using T4 polynucleotide kinase in the presence of gamma-ATP. For fluorophore detection, DNA and RNA oligonucleotides can be purchased with 5’ fluorophore linkages. We recommend terminating reactions with an equal volume of 2x stop buffer (see Recipes).
Notes
- Reproducibility
In our experience, extension of nucleic acids by Polθ is highly reproducible. However, we note that the precise amount of initial substrates extended may vary. For example, in some cases 100% of nucleic acid substrates are extended, whereas in other cases a small fraction (i.e., ~5-15%) of substrates are not extended. A 4-5 fold higher ratio of Polθ to nucleic acid substrates will usually allow for the majority of substrates to be extended. We note that the number of nucleotides transferred to the 3’ terminus of nucleic acids may vary. Thus, the final length of extended nucleic acids will not be identical for all molecules. The respective structures of canonical nucleotides and nucleotide analogs will also give rise to different terminal transferase efficiencies. For example, deoxyadenosine monophosphate is most efficiently transferred by Polθ (Kent et al., 2016). Other deoxyribonucleotides are somewhat less efficiently transferred by the polymerase (Kent et al., 2016). The initial nucleic acid sequence may also affect Polθ terminal transferase activity. Previous studies compare the efficiency of Polθ terminal transferase activity on different nucleic acid substrates and in the presence of various canonical nucleotides and nucleotide analogs (Kent et al., 2016). - Additional notes, technical tips and cautionary points
For optimal Polθ terminal transferase activity, we recommend storing the enzyme in buffer B (see Recipes) at concentrations ≥ 1 mg/ml in small aliquots at -80 °C and limiting freeze thaw cycles to 2-3 times. We note that oligonucleotides relatively short in length (< 10 nt) may not be extended as efficiently as those longer in length (> 10 nt). Oligos containing a high proportion of closely spaced guanosine bases, for example similar to telomere repetitive DNA sequences or those that form G quadruplexes, may exhibit a lower efficiency of extension by Polθ (Kent et al., 2016). As noted above, Polθ can also be used to modify double-stranded DNA, however, only 1-3 nucleotides are generally transferred to these substrates (Kent et al., 2016).
Recipes
- 1 M Tris buffer pH 8.2
Weigh 121.1 g of Tris Ultra Pure and add 800 ml of dH2O
Stir until dissolved, then adjust pH to 8.2 with HCl
Adjust final volume to 1 L with dH2O - Buffer A
20 mM Tris-HCl pH 8.2
0.01% NP-40
0.1 mg/ml BSA
10% glycerol - 1 M HEPES buffer pH 8.0
Weigh 238.3 g of HEPES and add 800 ml of dH2O
Stir until dissolved, then adjust pH to 8.0 with NaOH
Adjust final volume to 1 L with dH2O - Buffer B
50 mM HEPES pH 8.0
300 mM NaCl
10% glycerol
0.01% NP-40
5 mM DTT - 2x stop buffer
90% formamide
50 mM EDTA
0.03% xylene cyanol
0.03% bromophenol blue
Acknowledgments
This work was funded by National Institutes of Health grant 1R01GM115472-01 awarded to R.T.P. Competing interests: R.T.P. and T.K. filed a patent application about the use of DNA polymerase theta to modify the 3’ terminus of nucleic acids. The protocol described herein was adapted from previous studies (Kent et al., 2016).
References
- Black, S. J., Kashkina, E., Kent, T. and Pomerantz, R. T. (2016). DNA polymerase theta: A unique multifunctional end-joining machine. Genes (Basel) 7(9).
- Chan, S. H., Yu, A. M. and McVey, M. (2010). Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet 6(7): e1001005.
- Flett, F. and Interthal, H. (2013). Separation of DNA oligonucleotides using denaturing urea PAGE. Methods Mol Biol 1054: 173-185.
- Hogg, M., Seki, M., Wood, R. D., Doublie, S. and Wallace, S. S. (2011). Lesion bypass activity of DNA polymerase theta (POLQ) is an intrinsic property of the pol domain and depends on unique sequence inserts. J Mol Biol 405(3): 642-652.
- Kent, T., Chandramouly, G., McDevitt, S. M., Ozdemir, A. Y. and Pomerantz, R. T. (2015). Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat Struct Mol Biol 22(3): 230-237.
- Kent, T., Mateos-Gomez, P. A., Sfeir, A. and Pomerantz, R. T. (2016). Polymerase θ is a robust terminal transferase that oscillates between three different mechanisms during end-joining. Elife 5.
- Koole, W., van Schendel, R., Karambelas, A. E., van Heteren, J. T., Okihara, K. L. and Tijsterman, M. (2014). A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat Commun 5: 3216.
- Mateos-Gomez, P. A., Gong, F., Nair, N., Miller, K. M., Lazzerini-Denchi, E. and Sfeir, A. (2015). Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518(7538): 254-257.
- Sfeir, A. and Symington, L. S. (2015). Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? Trends Biochem Sci 40(11): 701-714.
- Summer, H., Gramer, R. and Droge, P. (2009). Denaturing URea polyacrylamide gel electrophoresis (Urea PAGE). J Vis Exp (32): 1485.
- Wood, R. D. and Doublie, S. (2016). DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair (Amst) 44: 22-32.
- Yousefzadeh, M. J., Wyatt, D. W., Takata, K., Mu, Y., Hensley, S. C., Tomida, J., Bylund, G. O., Doublie, S., Johansson, E., Ramsden, D. A., McBride, K. M. and Wood, R. D. (2014). Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet 10(10): e1004654.
Article Information
Copyright
![]() Hoang et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Hoang et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Hoang, T. M., Kent, T. and Pomerantz, R. T. (2017). Modification of 3’ Terminal Ends of DNA and RNA Using DNA Polymerase θ Terminal Transferase Activity. Bio-protocol 7(12): e2330. DOI: 10.21769/BioProtoc.2330.
- Kent, T., Mateos-Gomez, P. A., Sfeir, A. and Pomerantz, R. T. (2016). Polymerase θ is a robust terminal transferase that oscillates between three different mechanisms during end-joining. Elife 5.
Category
Molecular Biology > DNA > DNA labeling
Molecular Biology > RNA > RNA labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.